 Close
Close

Retinitis Pigmentosa (RP) is a peripheral retinal disease.
What is Retinitis Pigmentosa (RP) and is it really a bestrophinopathy?
RP is the commonest form of genetically inherited retinal degeneration, responsible for vision loss in approximately 1 in 4,000 people worldwide. Typically, there is a primary degeneration of rods followed by a secondary degeneration of cones. The age of onset of RP ranges from early childhood to up until the fifth decade of life.
The association of RP with the BEST1 gene was first described in 2009 with it thought to exhibit a dominant inheritance when there are mutations in BEST1. The exact prevalence of RP in those with mutations in BEST1 is currently not known. More recently it is thought that RP patients with BEST1 mutations may be misdiagnosed ADVIRC patients.
How does RP affect the eyes? What will the back of my eye look like in RP?
The typical primary degeneration of rods followed by the secondary degeneration of cones, results in patients presenting with initial night blindness and peripheral pigment deposits resembling bone spicules on fundus examination. Gradually, RP progresses from the periphery resulting in the appearance of more central retinal deposits, thin retinal vessels, and a pale optic disc with swelling of the macula also present, leading to the classic symptom of ‘tunnel vision’. Vision is greatly reduced. There may also be refractive errors such as myopia (short-sighted) and astigmatism (having a ‘rugby ball’ shape eye) which if uncorrected will affect vision.
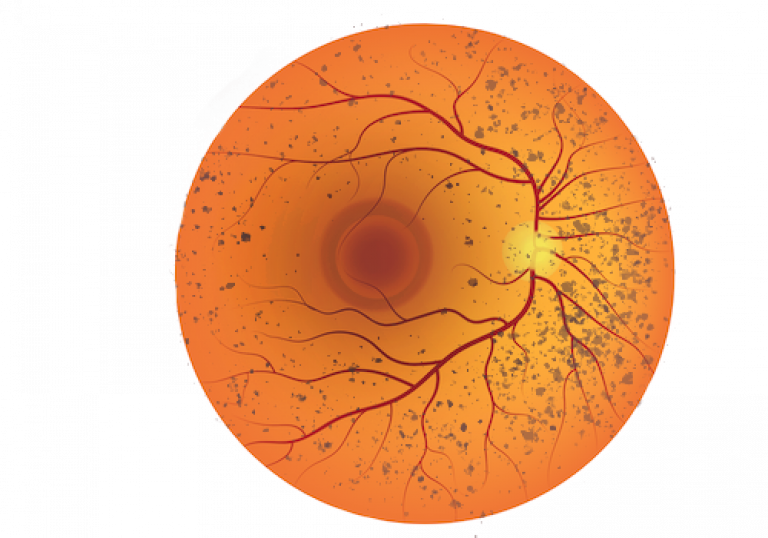
Which read-outs are unusual in RP?
OCT imagery can be useful to examine the retinal, identifying structural changes, fluid leakage and changes in the thickness of the layers of the retina.
Autofluorescence imaging can detect changes associated with autofluorescence in the eye, including the presence of increased pigmentation.
ERGs and EOGs will likely be abnormal in patients suffering from RP. Visual fields will be greatly impaired as patients in which the disease has progressed will have the characteristic ‘tunnel-vision’ field loss.
Are there any complications of RP?
In terms of complications for RP, the main complication is regarding the potential Vitamin A treatment for RP, which has been found to be toxic to some patients.
What are the treatment options for RP?
There is currently no medical or surgical gold-standard treatment for RP. In many cases current recommended treatment, such as Vitamin A supplements, only slows the disease down. Patients who suffer from refractive error, which is linked to RP, can be given prescription glasses or contact lenses. Patients with swelling and fluid accumulation can also be treated in hospital via eye injections or sometimes even different tablets or eye drops.
Research into treatments for RP is ongoing. Patients are encouraged to visit the many charities and support groups for bestrophinopathies.