 Close
Close

Theory & Modelling of Materials

Prof Jochen Blumberger
Our group carries out research to advance the predictive power of atom-scale computer simulations of materials and (bio)molecules. We also develop multi-scale models that bridge the gap between the atomistic and the experimentally relevant time and length scales. We have a keen interest to apply our methodologies to understand, at a fundamental level, the mechanisms of energy conversion processes in (opto-)electronic materials and to uncover structure-function relations that may help guide the design of improved materials. Recent projects include (i) the development of non-adiabatic molecular dynamics techniques for time-propagation of charge carriers and excitons in organic semiconductors (ii) the development of machine learning methodologies to accelerate ab-initio molecular dynamics with applications to solvation, redox- and electro-chemistry (iii) the development of density functional theory methodologies for the calculation of current-voltage characteristics of proteins in electronic junctions relevant for bioelectronic applications.
Prof Edina Rosta
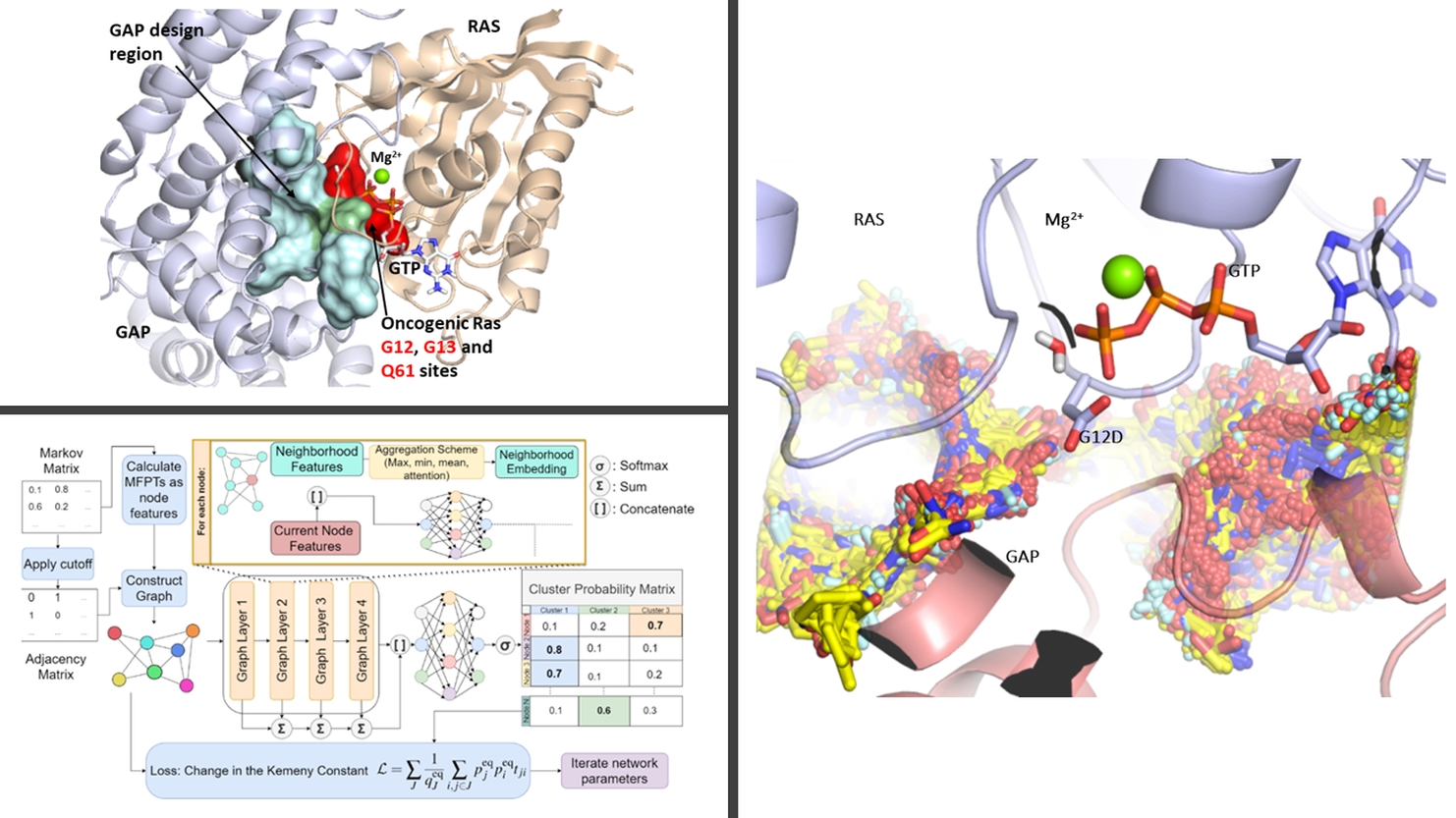
The Rosta research group focuses on how to enable computational design by understanding the catalytic power of enzymes using atomistic molecular modelling tools, including hybrid quantum mechanics/molecular mechanics (QM/MM) simulations. To quantitatively and accurately assess how enzymes achieve their extraordinary efficiency and specificity in performing chemical reactions, we develop and use modern enhanced sampling methods. Biased simulations are usually required to reach the relevant timescales of important biological processes using current simulation resources. We develop novel algorithms to calculate molecular kinetics in addition to free energies from biased molecular simulations using Markov chains defined on molecular conformational networks. Applications aim at understanding and molecular design concerning the most prominent chemical reactions of living organisms: phosphate transfer and cleavage.
Prof Alex Shluger
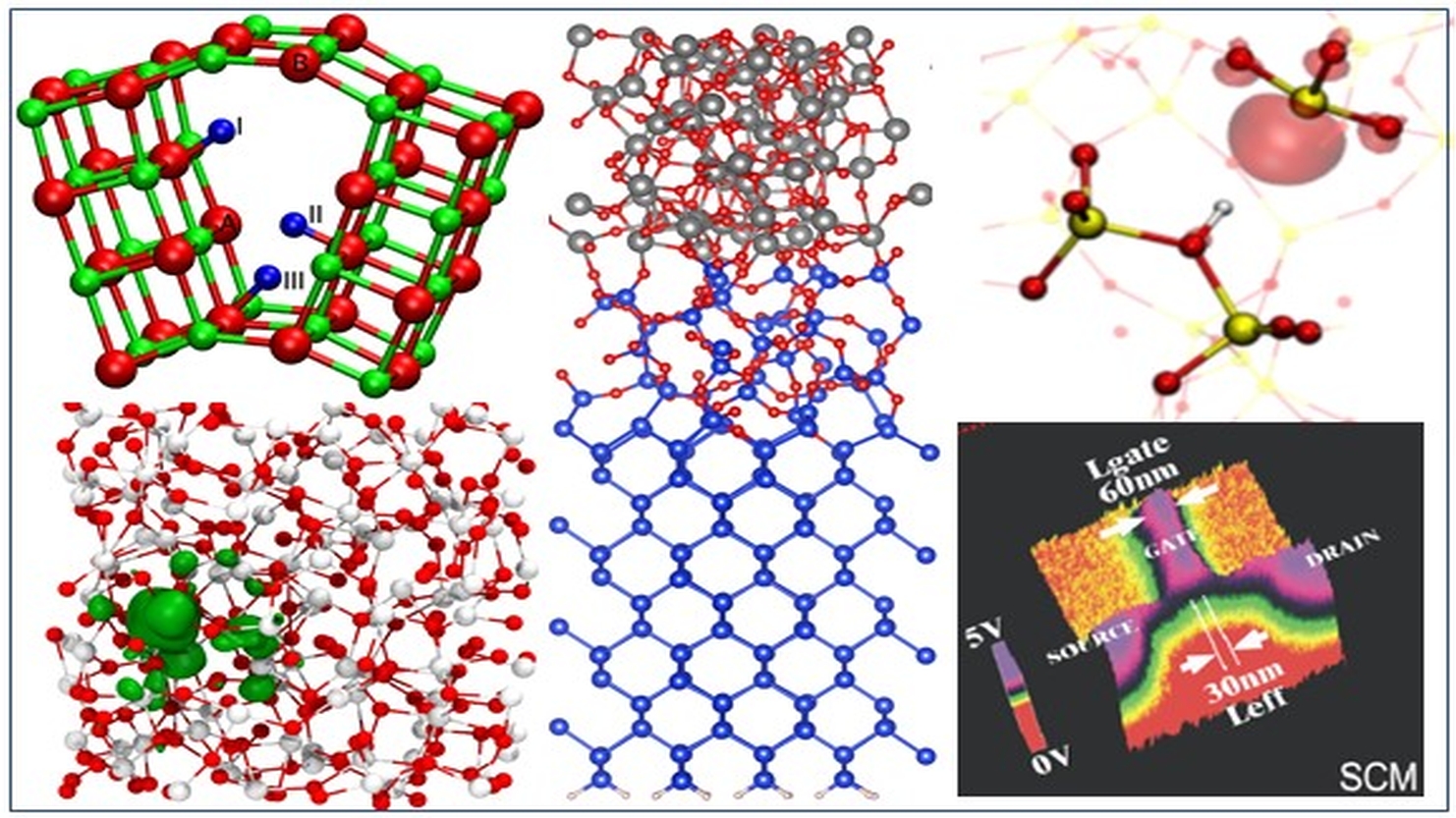
We develop and apply computational methods to study defects and defect related processes in solids and at interfaces. Of particular interest are metal/oxide/semiconductor and other hetero-structures used for novel memory and transistor devices. We investigate the mechanisms of resistance changes in thin layers of oxides, carbon nanotubes and 2D materials (bP, hBN, MoS2, WS2…), as well as their structural and electrical degradation and dielectric breakdown. Oxide materials include crystalline and amorphous MgO, SiO2, HfO2, Al2O3, TiO2, Ga2O3, ZnO sandwiched between Cu, TiN, Pt and Si electrodes as well as SiO2/H2O/WS2 hetero-structures. We are investigating the mechanisms of electron and hole injection from electrodes into these films, electron and hole trapping inside oxides, and defect creation under applied electric bias. We calculate probabilities of electron tunnelling through defect states determining leakage and breakdown current through dielectric films and consider the mechanisms of field and electron-induced defect reactions inside oxides, including hydrogen and other impurities.
Prof Ian Ford
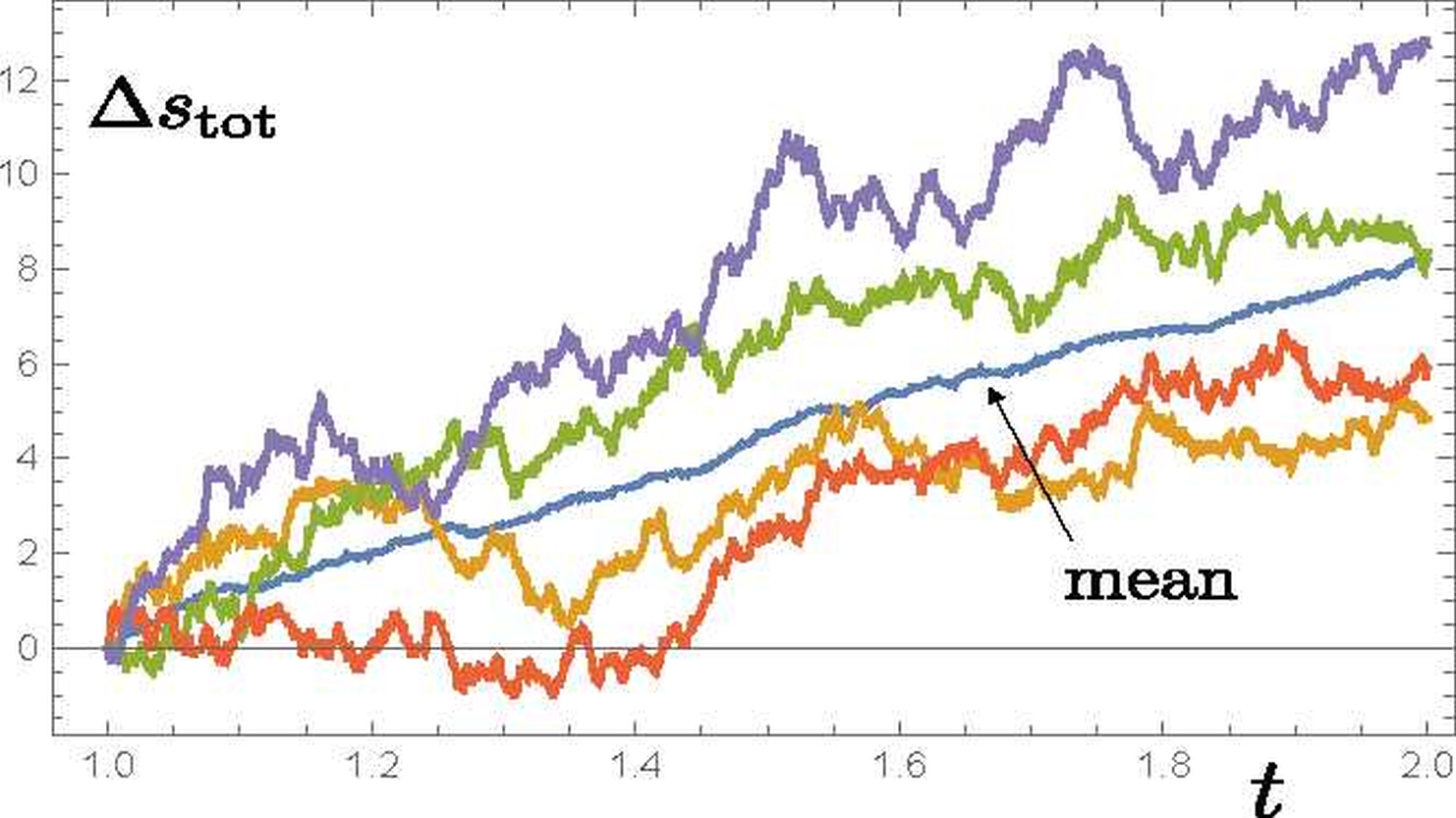
My research concerns the behaviour of systems out of equilibrium, such as phase transitions in molecular matter (for example water condensation, macromolecular structural selection, growth of crystalline materials), or quantum systems that are undergoing thermalisation or measurement. I am interested in the nature and meaning of entropy as a measure of the irreversibility of such transformations, and use statistical mechanics and stochastic thermodynamics to investigate various phenomena. The kinetics of crossing of thermodynamic barriers en route to equilibrium has been of particular interest, especially the nucleation of nanoparticles, such as aerosol formation from gaseous precursors.
Dr Frank Kruger
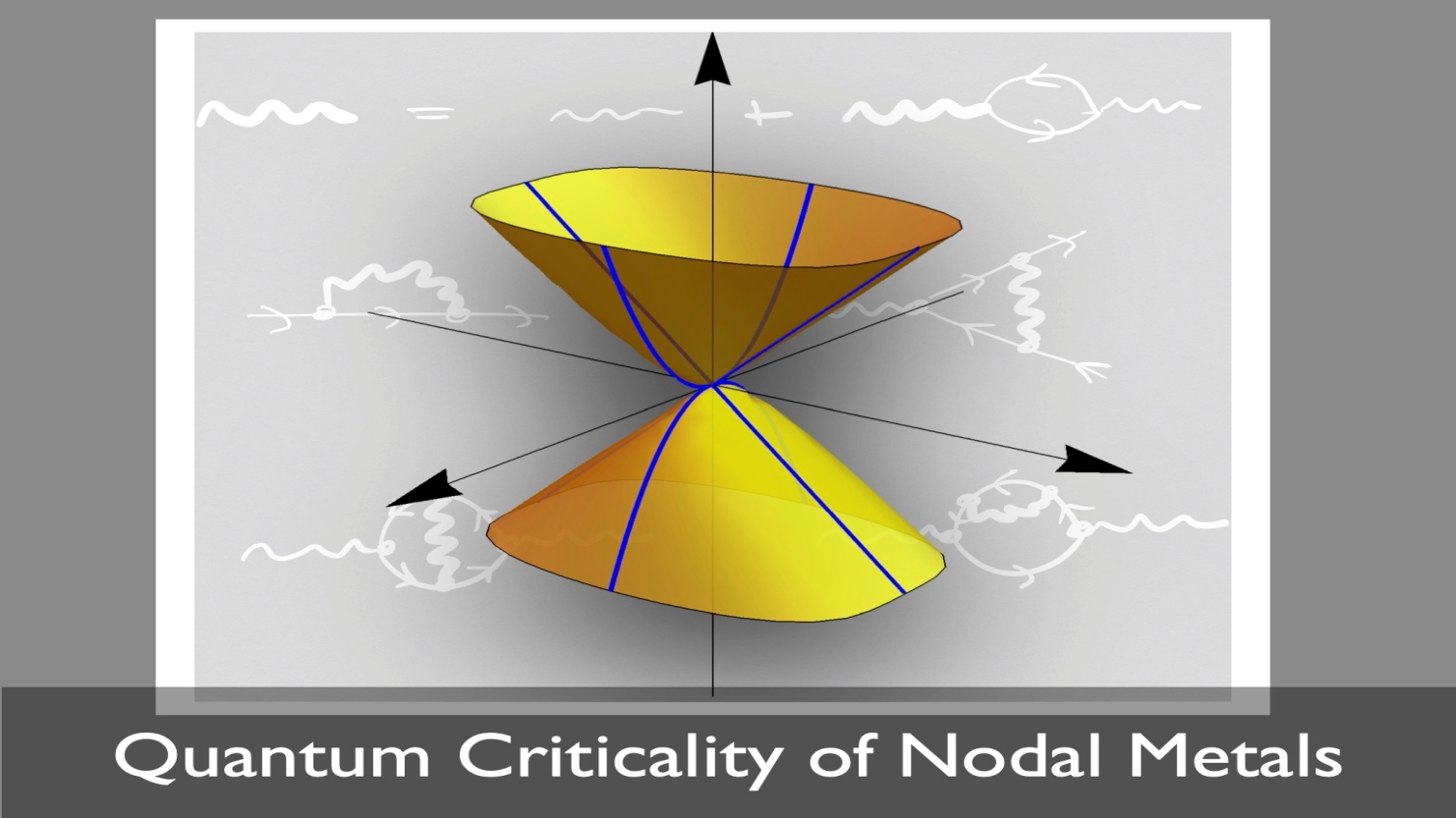
Our group investigates the formation of novel states of quantum matter in strongly correlated electron systems. We use analytical tools such as quantum field theory and renormalisation group calculations to understand the collective behaviour of electrons and the nature of quantum phase transitions between different phases of matter. More recently, we have started to study quantum spin liquids. These are highly frustrated magnetic materials in which strong quantum fluctuations prevent the formation of magnetic order down to absolute zero temperature. Remarkably, the spin degrees of freedom can break up or fractionalise into particles that satisfy fermionic quantum statistics. Although the emergent fermions in insulating quantum spin liquids don’t carry electric charge, they behave in many respects like interacting electrons in metals. In addition, they are coupled to gauge fields, resulting in even richer behaviours.

Prof David Bowler
We develop and apply methods to allow large scale calculations on the atomic and electronic structure of materials, concentrating on semiconductor surfaces, and more recently on ferroelectric materials such as PbTiO3. Standard approaches to these calculations are limited in the size of problem that can be studied to a few hundred atoms, but our methods allow calculations of up to 10,000 atoms with no approximation, and over 1,000,000 atoms with specific, well-controlled approximations. This allows us to address problems which are complex and interesting, such as the polarisation texture in thin films of PbTiO3 on SrTiO3 substrates.
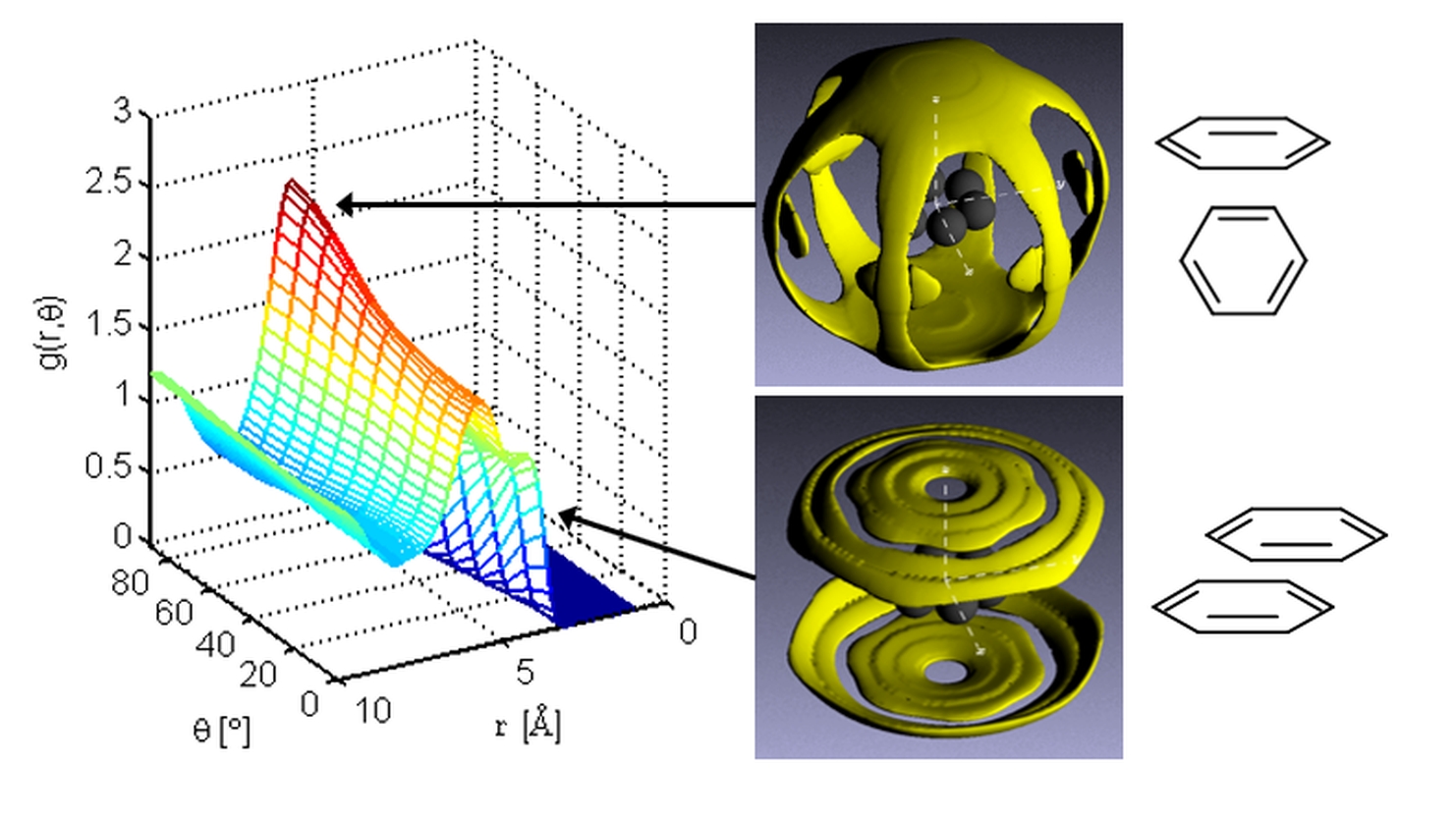
Prof Neal Skipper
Our research aims to understand and control the properties of a wide range of functional materials, by building up a picture of where the atoms are what the atoms do. We are particularly interested in the behaviour of liquids and complex fluids, and are investigating their role as solvents and electrolytes along with the nature of the fundamental interactions that take place at interfaces and in nanoscale confinement. These interactions are a key factor in many important biological and chemical processes. We use a variety of experimental and computational techniques in our research. In addition to our laboratories at UCL, we also make extensive use of international neutron and X-ray scattering facilities. For example the ISIS Neutron Source at the STFC Rutherford Appleton Laboratory, and the Institute Laue-Langevin in Grenoble.
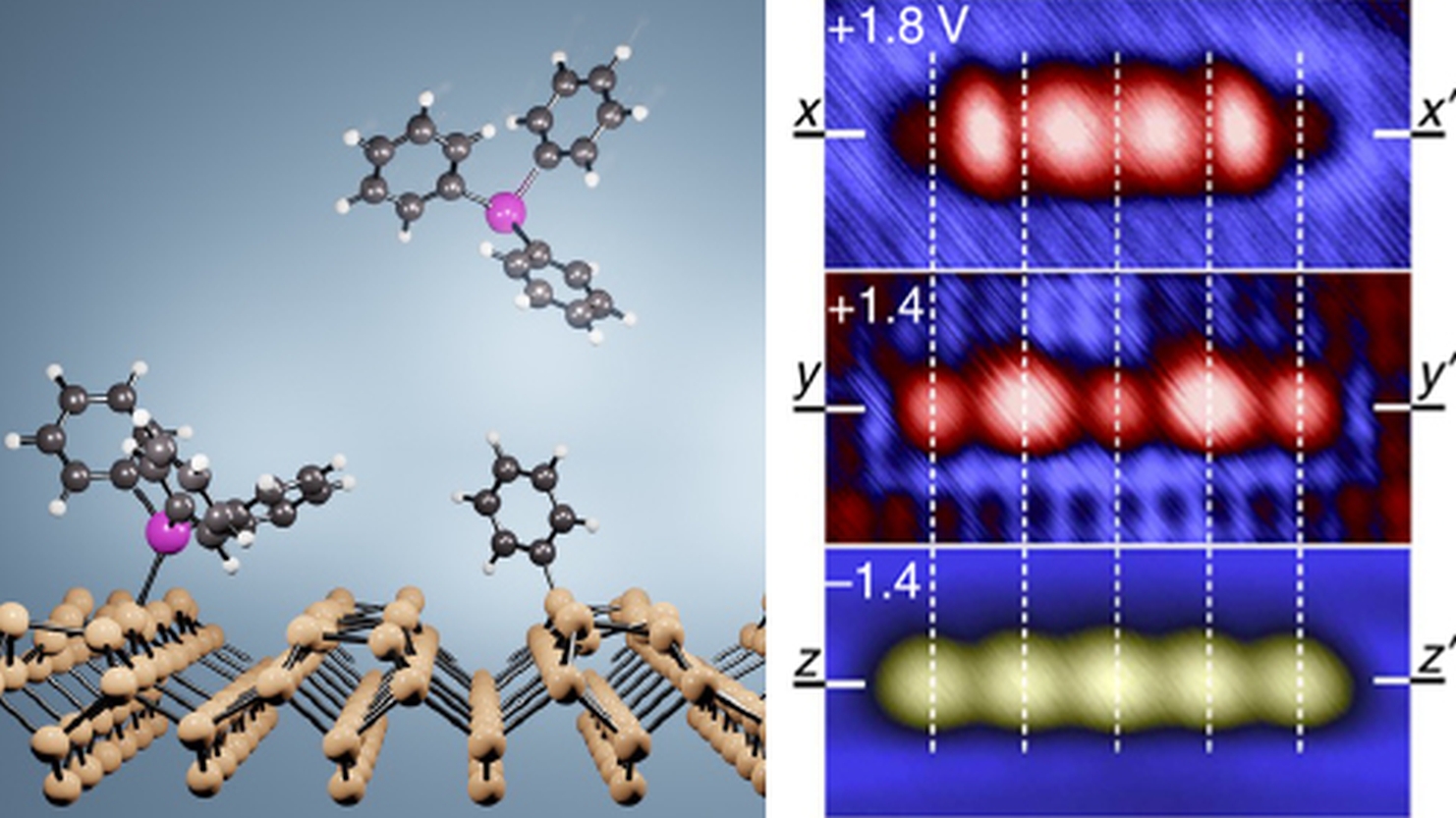
Dr Steven Schofield
Steven Schofield is an Associate Professor in the London Centre for Nanotechnologyand Department of Physics and Astronomy. He aims to understand and control matter at the atomic scale for fundamental discoveries in condensed matter physics and advancements in (opto)electronic devices and quantum technologies. His group employs atomic-scale fabrication and cryogenic-temperature scanning tunnelling microscopy to create and investigate nanostructures on semiconductor surfaces and two-dimensional materials. His team is developing artificial intelligence tools for efficient data collection and enhanced analysis objectivity. In parallel, a significant portion of their work is conducted through synchrotron-based photoelectron spectroscopy and theoretical approaches, including density functional theory. Students in his group are typically engaged in several, if not all, of these complementary approaches during their PhD project. Through this integrative approach, and with strong collaborations with national and international partners, they aim to develop a deep understanding of the quantum properties of matter at the atomic scale.

Dr Jonathan Breeze
Our research activities span many areas of condensed matter physics and quantum information science, but our core focus is the study of light-matter interaction, room-temperature solid-state masers and cavity quantum electrodynamics. We use computation tools such as density functional theory and quantum field theory to study the electronic structure, electromagnetic and vibrational properties of materials, Maxwell solvers to simulate and design devices such as cavities and waveguides (from microwave to optical frequencies) and numerical methods to investigate quantum spin dynamics. Our lab is equipped with pulsed electron paramagnetic resonance (EPR) spectrometers and optically-detected magnetic resonance (ODMR) spectrometers, which we use to characterise photo-excited spins in organic molecules such as pentacene and colour-centres in inorganic materials, such as nitrogen-vacancies in diamond.