Close
Close

Chemistry's Research into Covid-19
Prof Peter Coveney’s group in the Centre of Computational Science (CCS) is working with European and American collaborators on work that aims to accelerate the development of antiviral drugs for Covid-19 by modelling proteins that play critical roles in the virus life cycle in order to identify promising drug targets. The work is developing machine learning (ML), deep learning (DL) and artificial intelligence (AI) techniques to:
- identify and build accurate three-dimensional structural models of the SARS-CoV-2 proteome by closely integrating experimental structural and systems biology datasets,
- accelerate adaptive conformational sampling of the viral proteins to potentially identify novel binding sites/pockets that can be targeted by small molecules,
- rapidly filter, rank, and search for small molecules across widely available chemical libraries, and to integrate virtual screening (computational drug discovery techniques) techniques with experimental high throughput screening, and
- enable multi-scale, multi-resolution simulations of the SARS-CoV-2 viral envelope, and specific proteins.
Across various supercomputing facilities in the US and Europe, three collections of drug candidates are being screened for inhibitor activity:
- Known and licensed drugs for quick repurposing opportunities (e.g., DrugBank),
- Library of 100M known small molecules that are drug like (e.g., PubChem) and
- Large-scale libraries (e.g., Enamine, ZINC) with billions of compounds that could be manufactured quickly for testing.
Their primary initial targets are existing drugs that are currently in manufacturing pipelines and can be repurposed quickly. There are a number of compounds that are now being suggested by these models and we are making them available to community wet labs for experimental testing and screening.
The multi-level screening approach uses fast machine learning and docking methods to provide an initial ranking of drug compounds. This ranking is refined by using methods that take the protein response into account as well as the initial protein states. These methods include machine learning driven sampling as well as accelerated dynamics methods to sample the protein and protein-ligand complex phase space. The CCS group is carrying out more advanced binding free energy prediction calculations on thousands of compounds selected by this workflow. These computationally intensive calculations will be carried out the supercomputers Summit, Frontera, Longhorn, Theta, SuperMUC-NG, and Scafell Pike (at Hartree Centre).
The group are now seeking to identify promising inhibitors of COVID-19 targets through assessing the potential to reposition existing drugs in combination with machine learning alongside so-called deep-drive applications of molecular dynamics and artificial intelligence methods.

Image of SuperMUC-NG, supercomputer at Leibniz Supercomputing Centre of the Bavarian Academy of Sciences.

Photo by Oak Ridge Leadership Computing Facility at Oak Ridge National Laboratory. Summit is a next-generation IBM/NVIDIA supercomputer with an aggregate peak computer speed of over 200 PFLOP/S.
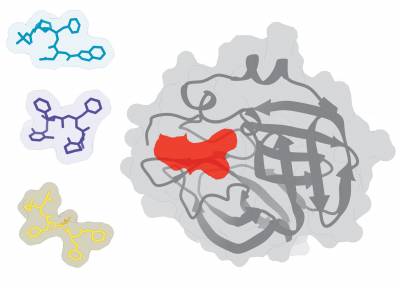
The main-protease of SARS-CoV-2 with 3 potential small-molecules that are approved for other targets.
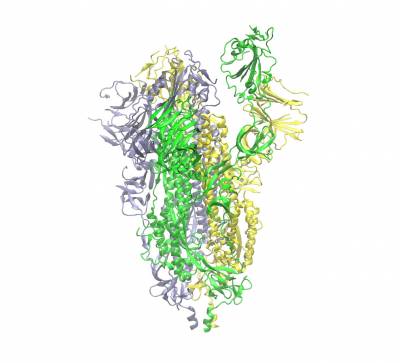
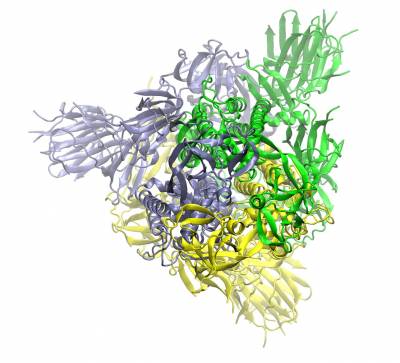
The trimeric spike protein (S-protein) protrude from the outermost envelope of the virus and has a receptor binding domain (RBD) that binds with the human ACE2 receptor allowing the viral genes/proteins to enter a human cell beginning its reproductive cycle. The binding process involves opening up one of the three chains of the S-protein (hence the partially opened conformation shown). It is one of the targets for which inhibitors are being developed. Human antibodies also target this protein.
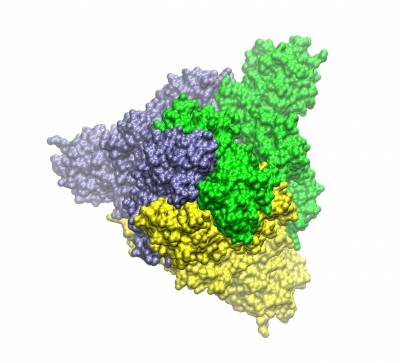
Trimeric spike protein of SARS-CoV2 (the three chains shown in different colours). Top view of the closed conformation
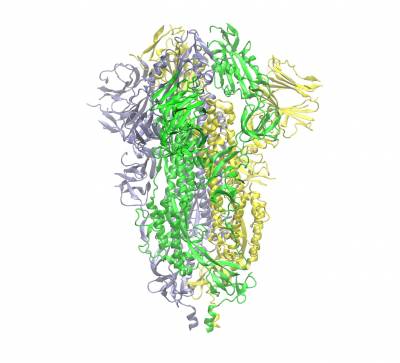
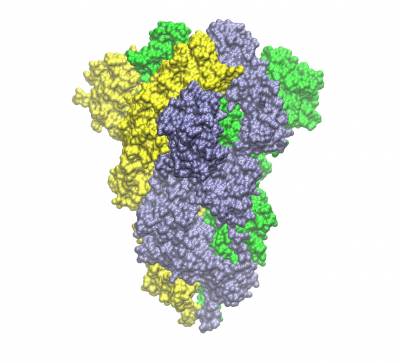
Trimeric spike protein of SARS-CoV2 (the three chains shown in different colours). Side view of the closed conformation