 Close
Close

Genetics and Cell Biology of Sarcoma
Group Leader: Professor Adrienne M Flanagan
BCh, BAO, LRCPI, LRCSI, FRCPath, PhD, FMed Sci, NIHR Senior Investigator, OBE, Hon Doctorate RCSI
Our Research Focus
This team studies the genetics and cell biology of sarcoma – tumours of bone and soft tissue. The focus is to refine the classification of bone and soft tissue tumours using molecular markers and to identify biomarkers for prediction for both response to therapies and clinical outcome.
Sarcomas
Sarcomas are malignant tumours of the musculoskeletal system (bone and soft tissue) representing less than 2% of all cancers. However, the centralisation of treatment of patients with these tumours in the London Sarcoma Unit allows basic and clinical research to succeed. This gave us the opportunity to biobank these rare tumours and over the last decade, we have contributed to the refinement of the classification of primary bone tumours through the discovery of new genetic alterations which define specific tumour types (Henderson S. et al., 2005, Amary F. et al., 2011, Behjati S. et al., 2014).
Now, we are exploiting the molecular profiles of these tumours in two ways:
- by developing biomarkers and employing them as clinical tests to provide patients with more accurate prognoses and to predict if tumours are sensitive to conventional chemotherapy and new targeted treatments.
- by interrogating the tumours by applying state of the art technology using in vitro models of disease to study the genetic alterations, alone and in combination, to determine their significance in terms of tumour growth and behaviour, but also to determine how these induce resistance to conventional chemotherapy and new targeted treatments.
Sarcoma and the 100,000 Genomes Project
The 100,000 Genomes Project has demonstrated that genomics can play a pivotal role in improving diagnosis and treatment for patients with sarcoma. At least 1,000 patients with sarcoma have been recruited to the 100,000 Genomes Project, 600 of whom were recruited at the Royal National Orthopaedic Hospital as part of the London Sarcoma Service.
Already we have seen changes in practice in pathology laboratories as a result of this project. Preliminary analysis of the genomes shows that more accurate diagnoses can be provided, and patients with germline alterations can be identified more readily. Best of all, this project has resulted in a more collaborative and comprehensive research strategy for this rare disease. More about Genomics England Clinical Interpretation Partnership (GeCIP) here.
As of early 2020, all sarcomas or tumours suspected of being sarcoma can be submitted to NHS England for whole Genome Sequencing and these genomes can be added to the profiles of tumours being analysed as part of the 100,000 Genomes Project.
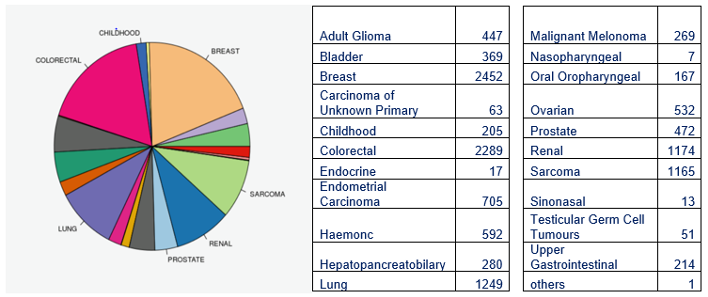
Whole Genome Sequencing results returned by tumour type from Genomics England (n=16,370).
Sarcoma represents no more than 2% of all cancers but due to the commitment of NHS staff across the Genome Medical Centres, the numbers of sarcoma genomes analysed are greater than all cancers except breast and colorectal.
Genetics and Cell Biology of Sarcoma in Depth
Find out more about the research and development of our research and our team



Summer Studentships for Students Studying Medicine
The team welcomes, every summer, medical students (first and second year) who are interested in undertaking a short research project. This is a great opportunity to put into practice techniques which are learnt during their studies and being exposed to the research environment while attending lab meetings and conferences.
Studentships are available, kindly sponsored by Shionogi and The Pathological Society.
To enquire email Catherine Storm: catherine.storm.14@ucl.ac.uk
Last updated: April 2020

