 Close
Close

Only once the regulators approve the application can clinical trials begin.
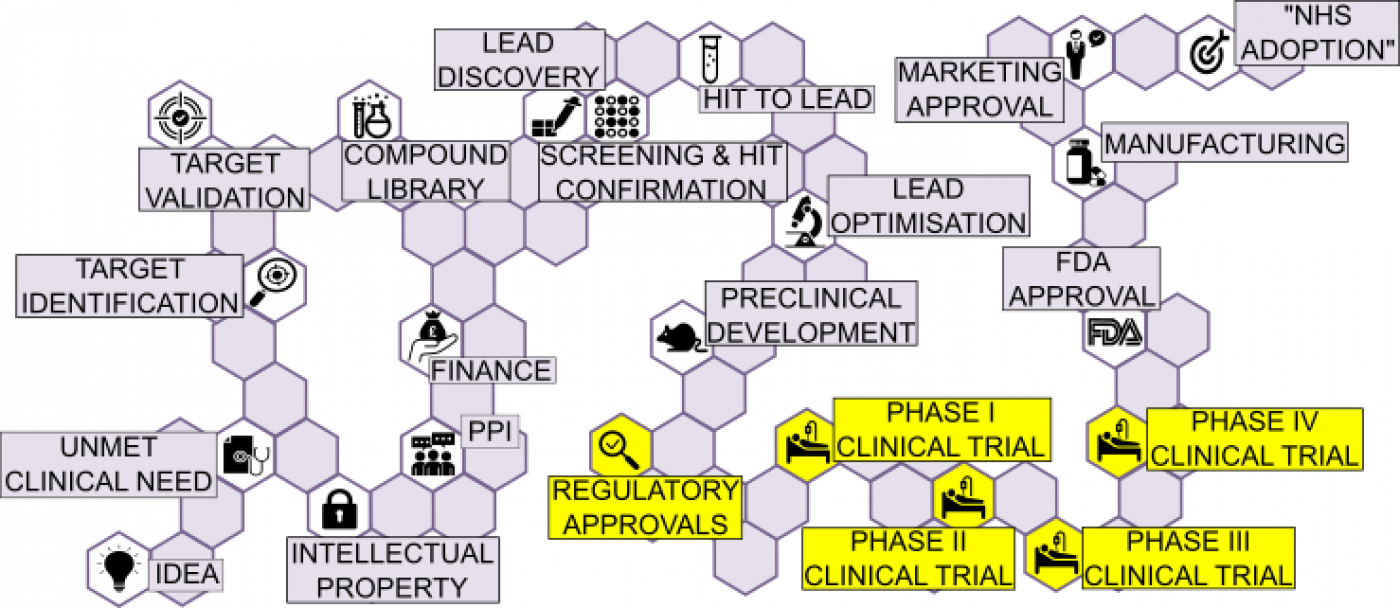
 REGULATORY APPROVALS:
REGULATORY APPROVALS:
Once the preclinical compound is selected, the data is collated to support an approval of a Investigational New Drug (IND) application. This application is given to a regulatory body – The Medicines and Healthcare products Regulatory Agency (MHRA)/the Food and Drug administration (FDA), so that the compound can move forward into human clinical trials.
The application must include:
- Animal study data and toxicity
- Manufacturing information
- Clinical protocols for the proposed human trials
- Data from any prior human research
- Information about the principal investigator(s).
Only once the MHRA/FDA approves the application can the clinic trials begin. It is rare that the IND application are refused at this point but MHRA/FDA can suggest improvement. This is because a lot effort, time and money is used to get the compound this point.
UCL Support:
The Joint Research Office (JRO), Clinical Trials Unit and Translational Research Office (TRO) support developers in seeking local, national and international approvals (Ethics, HRA, MHRA, R&D). Support can also be sought from local PPI leads within the JRO.
More information:
 CLINICAL TRIALS:
CLINICAL TRIALS:
The clinical trials are phased studies on volunteers to observe and collect data on the safety and efficacy of the compound.
Each phase of clinical trial is described below:
Phase I - This phase of clinical trials are tested on approximate 10-100 healthy consented volunteers. This non- blinded trial assesses the safety and tolerability of the drug candidate, to determine any adverse effects. As well as this the pharmacodynamics and ADME are monitored, therefore observing the time it take for the drug to be metabolised and excreted from the body. Trials must be compliant with Good Clinical Practice (GCP).
Phase II - This phase of clinical trials are tested on approximate 50-500 patient expressing the targeted disease. These studies are carried out mainly to see the effectiveness of the drug in patients. Although other data can also be collected from this phase like studying the dose- response relationship and determine a dosing regimen. In addition the safety of the drug within these sets of volunteers are also monitored. This phase clinic trials are randomised and are either a single or double blind trial. Having controlled trials where a placebo is given instead of the drug in testing, helps compare the data.
Phase III - This phase of clinical trials are tested on approximate 500-3000 or more patients that have the targeted disease. These trials take place over many sites and over a wide verity of patients so that the results can be compared to existing treatment or standard of care of the targeted disease. As well as comparing the efficacy with other drugs and the effect of different dosage. The trials in the phase are more complex and require a lot of funding to be able to be carried out. These trials are randomised, controlled double blind trials. If the outcomes from all the trials are positive, the data is then gathered and New Drug Application (NDA) is submitted to the MHRA/FDA for approve the licence of the drug.
Phase VI - This is where the follow up studies are carried out, to detect unusual or long- term side effects across a large population to look for patterns.
UCL Support:
The Comprehensive Clinical Trials Unit at UCL (Comprehensive CTU) collaborates with researchers to design, conduct, analyse and publish clinical trials and other well-designed studies.
The CCTU is a registered CTU and coordinates multi-centre clinical trials (i.e. has overall responsibility for the design, development, recruitment, data management and analysis of trials) with robust systems to ensure the highest quality standards of conduct and delivery of clinical trials.