 Close
Close

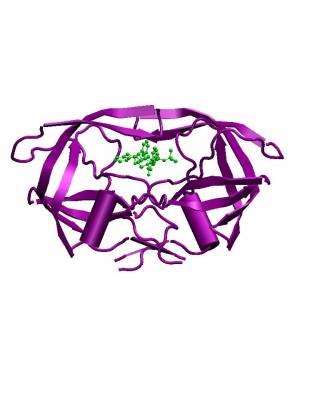
HIV-1 Protease is an aspartic protease and is one of the key enzymes in HIV, essential for the maturation of new infectious virions emerging from the host helper T-Cell. It is responsible for the cleavage of two HIV polyprotein chains into several distinct proteins including the protease itself, all of which are found in the mature virus.
Structurally it consists of an often rotationally symmetric dimer with monomoer subunits of 99 amino acids, resembling a "bulldog's face". The functionally active site consists of two aspartic acid residues (one from each monomer) bound by connected residues and by a pair of highly mobile "flaps".
The important role of HIV-1 protease in the life cycle of HIV has made it a crucial target for developing inhibitors that impede the progress of the virus within the body. To this effect, several HIV protease inhibitors (HPIs) have been developed to obstruct the active site of the protease and thus impede the cleavage of HIV polyproteins. However due to the highly mutational characteristics of the retrovirus, drug resistant strains frequently emerge to invalidate the efficacy of developed PIs.
We are studying the differential dynamics of several muational strains of HIV protease along with the wild type, both as an apo-protease and complexed with developed PIs, through the application of large scale molecular dynamics. We hope to elucidate the difference in dynamical behaviour of wild type with mutants and determine how this contributes to the differences in binding affinities between different strains. We also hope to use themodynamic integration techniques to calculate difference in exact binding affinities between different strains.