Close
Close

We have a diverse programme of pre-clinical and clinical research programmes dedicated to the development of new interventions for a broad range of sight-loss conditions.
Our Research
- Clinical Studies Update
Achromatopsia (Gene: CNGB3 and CNGA3)
An inherited, but not progressive condition affecting the cone (daylight) photoreceptors responsible for daytime vision. Achromatopsia is characterised by low vision, sensitivity to light and total colour blindness.
- We have previously shown it is possible to correct vision with gene therapy in a mouse model of CNGB3-associated achromatopsia (Carvalho et al. 2011)
- We have secured funding and manufactured a gene therapy for CNGB3-associated achromatopsia which is now in an early phase human safety trial (NCT03001310)
- We have plans to conduct a similar early phase human safety trial for CNGA3-associated achromatopsia in the future
Leber Congenital Amaurosis Type 2 [LCA2] (Gene: RPE65)
An inherited condition affecting the retinal pigment epithelium (RPE) at the back of the retina which supports and nourishes the photoreceptors. Loss of RPE function results in loss of photoreceptors leading to a progressive loss of vision.
- We previously completed a world-first early phase human safety trial of gene therapy for RPE65-associated LCA, which demonstrated that gene therapy is well-tolerated with no unexpected side effects and that some patients do experience improvements in their ability to navigate in dim light but this declines over time (Bainbridge et al. 2015)
- Our evidence suggests that current gene therapies may not fully meet the demand for RPE65 in humans. Therefore, we have developed a more potent gene therapy for RPE65-associated LCA which may provide longer-lasting benefits and might also offer improvements in daytime vision (Georgiardis et al. 2016)
- We have secured funding and manufactured this more potent RPE65 gene therapy which is now in an early phase human safety trial (NCT02946879)
X-Linked Retinitis Pigmentosa (Gene: RPGR)
An inherited form of progressive vision loss that affects both rod and cone photoreceptors. Caused by a faulty RPGR gene on an X chromosome, X-linked RP affects mainly males.
- Together with collaborators in Boston and Edinburgh we have previously shown it is possible to correct vision with gene therapy in a mouse model of RPGR-associated X-linked RP (Pawlyk et al. 2016)
- We have secured funding and manufactured a gene therapy for RPGR-associated X-linked RP which is now in an early phase human safety trial (NCT03252847 )
Leber Congenital Amaurosis Type 4 [LCA4] (Gene: AIPL1)
An inherited condition affecting all photoreceptors, leading to a rapid degeneration of the retina and loss of vision in the first few years of life.
- We have previously shown it is possible to correct vision with gene therapy in a mouse model of AIPL1-associated LCA (Tan et al. 2010, Sun et al. 2011)
- Despite the early-onset of photoreceptor loss and vision decline in people with LCA4, we have identified that there may be a window of opportunity to treat younger children (Aboshiha et al. 2015)
- Thanks to generous support and fundraising work by donors we have manufactured and safety tested an AIPL1 gene therapy. Children under three years of age with a confirmed genetic diagnosis of LCA caused by AIPL1, are thought to be those who may experience a potential benefit. For more information please contact Prof Michel Michaelides at michel.michalides@moorfields.nhs.uk
Positioning in Macular hole Surgery (PIMS) Trial
Macular holes are spontaneous breaks in the retina, most common in older women, leading to blurred and distorted central vision. They can be repaired surgically by injecting a temporary bubble of lighter than air gas into the eye which sits on the hole causing it to lay flat and helping it to seal.
- We have previously demonstrated that following surgery, rates of closure for large macular holes can be improved when patients spend several hours a day lying face-down (Lange et al. 2012)
- Lying face-down after surgery is good medical practice but can be burdensome for patient’s health and quality of life compared to sitting in a less burdensome face-forward position, but there is only limited evidence to support the face-forward option versus the face-down position (Solebo et al. 2011)
- We are now conducting a clinical trial to determine conclusively whether patients can get the same surgical outcomes from sitting face-forward as lying face-down (SRCTN1241059)
Aflibercept for Macular Oedema with Underlying Retinitis pigmentosa (AMOUR) Study
Retinitis Pigmentosa is the name given to a family of inherited sight loss conditions which lead to progressive loss of vision. A complication of retinitis pigmentosa in some patients is oedema (water-logging) underneath the macula, the part of the retina responsible for central vision, that causes further damage and vision loss. A treatment called Aflibercept may be effective in treating macular oedema (Strong et al. 2016)
- We are now conducting a clinical trial to determine whether Aflibercept can be used safely to treat macular oedema in people with retinitis pigmentosa (NCT02661711)
Stargardt disease
A form of inherited, progressive central vision loss that leads to a loss of photoreceptors. It often arises in early childhood but there are later onset forms of the condition which result in less severe loss of vision.
- In 2012, we formed a partnership with Advanced Cell Technology (also known as ACT or Ocata Therapeutics – now part of Astellas Pharma) to conduct Europe’s first-ever safety trial using stem cell-derived retinal cells to repair the macular in people with advanced Stargardt disease. The results from this study have demonstrated that 12-months following intervention, the stem cell-derived retinal cells are tolerated by the patients and there are no unexpected side effects. Future research would be required to demonstrate any long-term benefits for vision (Mehat et al. 2018).
- We are also involved in studies monitoring the natural progression of various forms of Stargardt disease to develop measures to inform future clinical trials (Progstar)
- Gene Therapies
Many inherited eye conditions are currently untreatable because they are caused by mutations (mistakes) in our own DNA. Our DNA is arranged into genes. Genes are instructions which regulate the normal functioning of our cells and organs. Sometimes, when mutations arise in one of the many genes responsible for normal visual function, this can lead to diseases that can run through a family.
The aim of gene therapy is to use our own DNA as a medicine to correct these harmful mistakes in our genes. Using harmless viruses, called vectors, we can deliver ‘normal’ copies of genes into the eye to correct for the damaged gene. The eye is a good candidate for gene therapy as it is relatively protected from the body’s powerful immune responses against foreign material like viruses that could otherwise limit the benefit of gene therapy.
However, gene therapy is highly specific to the genetic cause of condition so it is likely that every inherited eye condition, amenable to gene therapy, would need its own therapy and each individual would need to undergo genetic testing to make sure they were getting the appropriate therapy.
Research in this area shows exciting promise for prospective treatments in the future. We are currently investigating the following approaches:
Gene Replacement
When people describe gene therapy this is the approach they are usually referring to. Most clinical trials of gene therapy conducted to date are using this gene ‘replacement’ strategy where a normal version of the affected gene is delivered into the cells where it is needed, to compensate for the deficiency caused by the mutation.
Gene replacement is a suitable option when a patient has a condition resulting from a mutation in a single, relatively small gene.
Gene Editing
Gene editing technology aims to cut out the damaged part of a gene from a person’s own DNA and replace it with the normal part of the gene. With this technique, scientists can potentially develop effective therapies for much larger genes than current gene therapy techniques allow for and potentially expand the range of conditions that might benefit.
Right now, gene editing isn’t very efficient and can result in collateral damage to normal genes and this could cause further harm. Current research is aiming to make this technology more efficient and precise enough that we can consider using it medically.
Optogenetics
Optogenetics aims to introduce new genes into the retinal cells to change the way they respond to light.
This type of approach might be beneficial in advanced retinal degeneration where all the photoreceptors have been lost; enabling us to make cells which are not light sensitive now react to light and act like a photoreceptor.
This technology is in early stages of pre-clinical development.
- Cell Therapies
At the advanced stages of many inherited retinal conditions, termed end-stage disease, most of the photoreceptors and support cells of the retina have degenerated and been lost. In this instance, gene therapy is no longer an option as there are no cells left to treat.
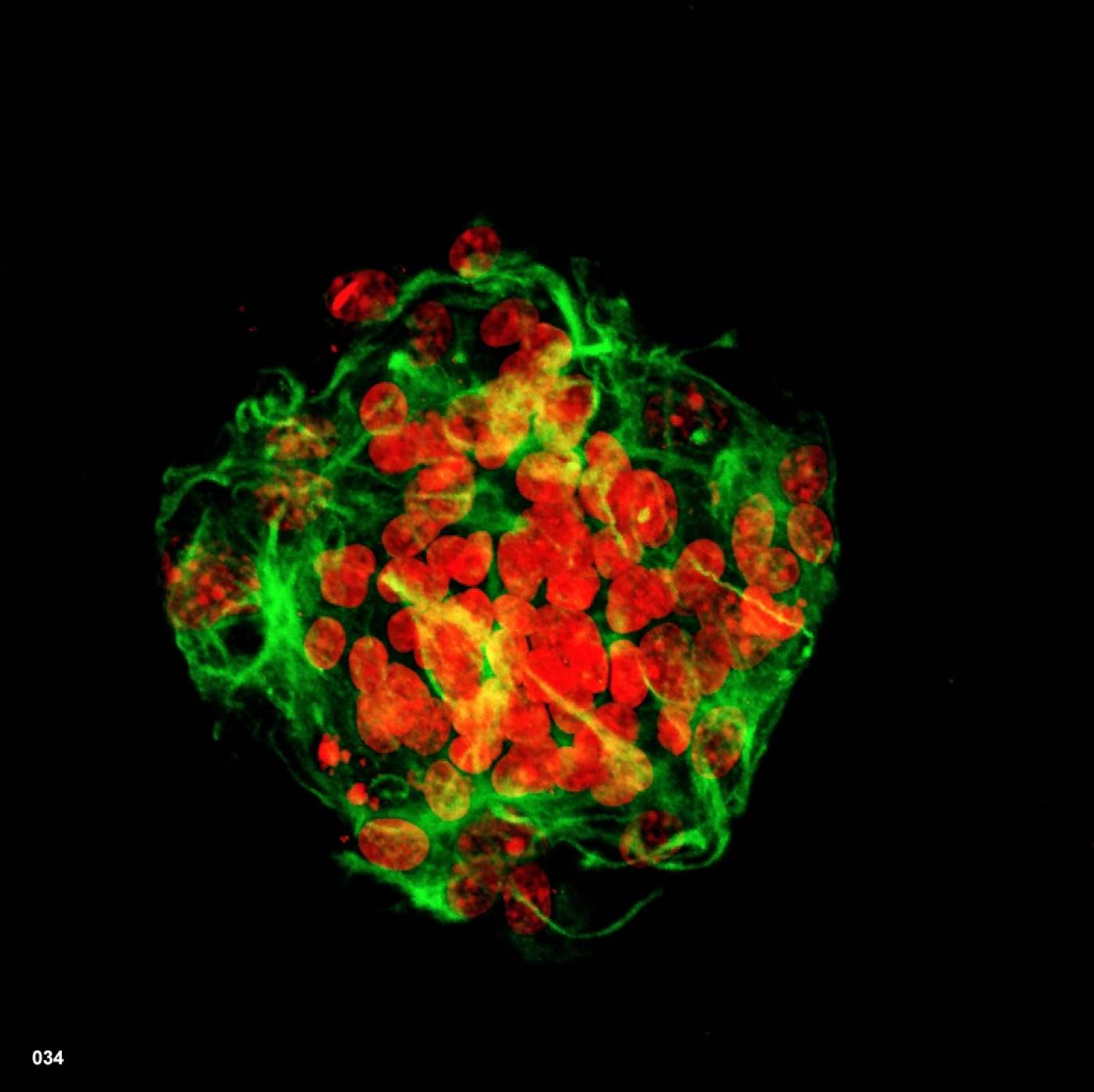
Currently, it is not possible to replace the eye or even the entire retina but it has been shown in many animal models of inherited retinal disease that it is possible to transplant patches of healthy donor cells into the eye. Recent findings from our group and others show that donor cells transplanted into these eyes appear to form new connections with the remaining retina and can drive visual function.
Stem cells represent a valuable source of new cells for transplantation. Stem cells are undifferentiated: cells which have not yet committed to becoming a specialised cell-like a skin cell, heart cell or a retinal cell. Stem cells are very good at replicating themselves and we can grow them up in the laboratory in relatively large numbers. By exposing them to the right combination of ‘factors’ we can make stem cells develop into a particular type of cell from a broad range of specialised cells (this ability is called pluripotency) suitable for transplantation including photoreceptors and retinal pigment epithelium. We use stem cells derived from two main sources 1) cells from fertilised eggs and 2) cells from adult tissue e.g. blood and skin.
With further development, transplantation of stem cell-derived retinal cells offers a promising approach for the treatment of advanced retinal degeneration.
Differentiation and transplantation of stem cells
Across several landmark studies, we have shown that in end-stage disease immature photoreceptor cells integrate with the host retina when transplanted into animal models of inherited retinal degeneration, provided they are at the correct stage of development.
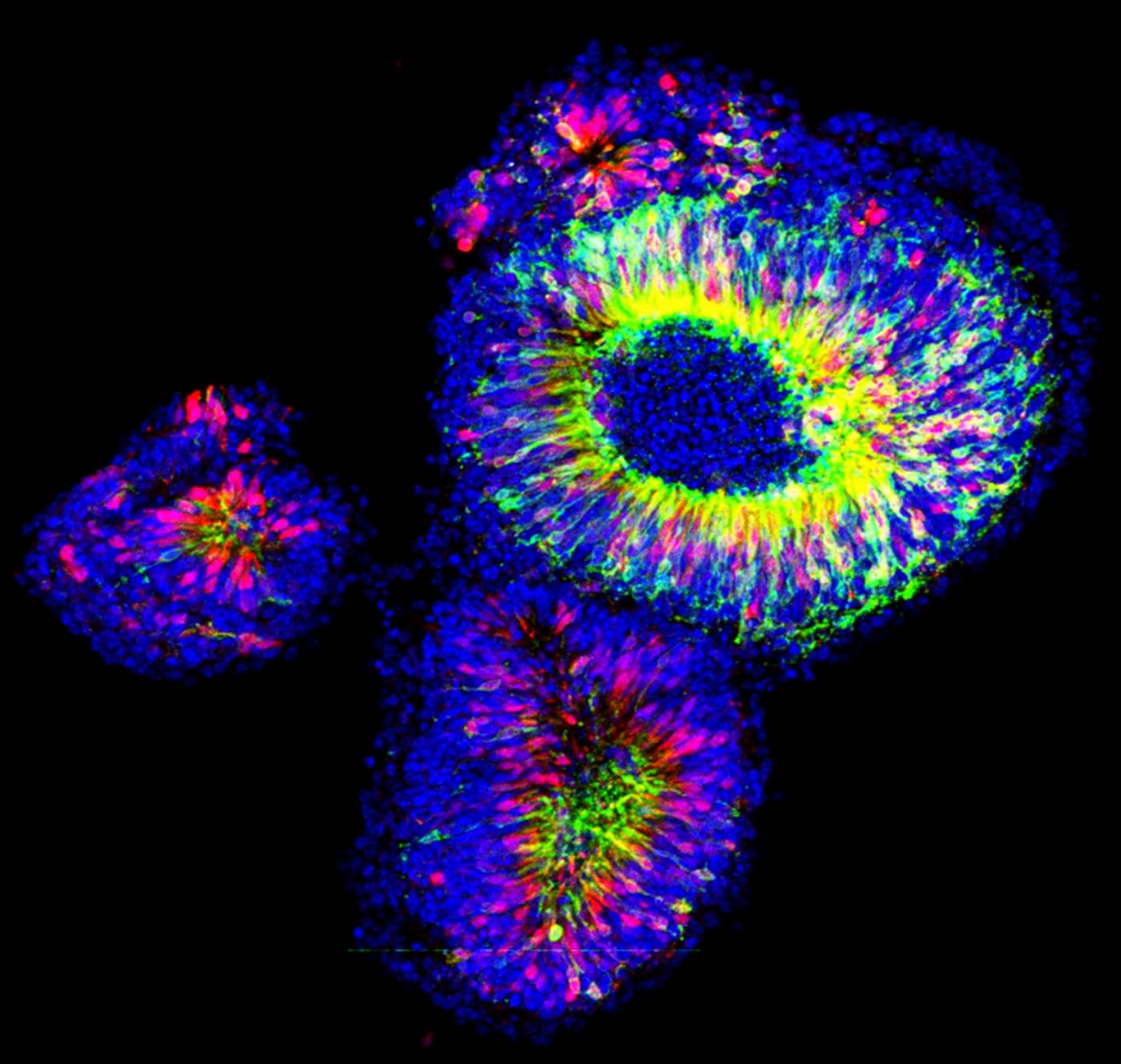
It is not possible to obtain immature photoreceptor cells at the right stage of development from humans, as this stage of development corresponds to the second trimester of pregnancy. In 2013, we further optimised a new ground-breaking stem cell technique from Japan to generate a ‘mini retina’ in a dish to give us sufficient cell numbers at the right stage of development and suitable for transplant (3D cell culture).
We are currently working to improve the success rate of cell transplants and are adapting these proven techniques to apply these technologies to human cells with the aim that we can develop cells suitable for human transplantation in the future.
Good Manufacturing Practice (GMP)
GMP refers to the standards that new medicinal projects must conform to before they can be used in people. We are currently working on a GMP compliant process for our stem cell research. Our aim is to develop a cost-effective and scaled up version of our current research methods to produce stem cell-derived retinal cells, that can be stored long-term for transplant into people.
Induced Pluripotent Stem Cells
It is now possible to take a tissue sample from a person and, in the laboratory, make the cells of the tissue revert back to a stem cell-like state called an induced pluripotent stem cell (iPSC). By exposing iPSCs to combinations of the right ‘factors’ we can encourage them to form other cells e.g. we can take iPSCs derived from human blood and grow these into 'mini retinas' in a dish.
Using iPSCs, it might be possible to reduce the chance of rejection after transplantation. For example, a blood sample from a patient with an inherited retinal dystrophy could be used to create iPSCs, which could then be treated with gene therapy to correct the disease-causing mutation. The iPSCs could then be used to grow new photoreceptors for transplantation. Once transplanted there should be virtually no chance of the new cells being rejected because they are from the same person.
Growing iPSCs derived from a patient’s tissue sample can also be a powerful research tool to study inherited retinal diseases more closely. By growing retinal cells derived from these iPSCs we can study the retina in the early stages of development and better understand at what point something goes wrong and how this progresses. These new insights into the mechanisms of the disease may reveal new approaches to treating these conditions. As we develop new treatments for inherited retinal diseases, these iPSC retinas provide more human-like models to test therapies on so we can better understand how these might affect a patient.
Material Transfer
In 2016 we and other groups published independent studies showing that a new and previously unknown mechanism of interaction between transplanted photoreceptors and the photoreceptors of the recipient retina, called Material Transfer, plays a major role in the rescue of visual function.
In Material Transfer ‘packages' of genetic and cellular molecules, such as RNA and/or protein(s) from donor cells are released and then taken up by the degenerating photoreceptors. This process appears to provide degenerating photoreceptors with sufficient protein(s) (or instructions to produce these proteins) to drive a restoration of visual function.
More immediately, cell transplantation is likely to be of benefit to those patients with end-stage disease. However, should we be able to determine the underlying mechanism, then material transfer may represent an exciting new therapeutic approach in earlier stages of degeneration to deliver functional biological molecules into degenerating photoreceptors and rescue visual function.
- Mechanisms of Disease
There isn’t a single mechanism by which all eye diseases work. There are many different genes required for normal visual function, each one with a different role working at a different point in the visual pathway, which is very complex with lots of overlapping processes. Therefore, a mutation in two different retinal genes may affect vision very differently; mutations in AIPL1, for example, lead to a very rapid loss of vision in early childhood in people with LCA4 while people with Achromatopsia who have mutations in CNGB3 or CNGA3 retain much more of their useful vision.
Inherited retinal diseases may have secondary impacts as well, causing other pathways of the body to act inappropriately as they try to function or compensate for the knock-on effect of the disease-causing mutations.
Oxidative stress
The retina has the highest requirements for energy of any tissue in the body and needs a rich supply of oxygen to function normally. In retinal diseases like retinitis pigmentosa, where the cells of the retina degenerate, as cells are lost the oxygen supply doesn’t reduce accordingly. The remaining cells are put under increased stress as they try to manage the various toxic products (reactive oxygen species) that arise from the abnormally high oxygen levels they now experience.
The accumulation of these toxic by-products can lead to further cell stress and death. We are investigating the roll of oxidative stress in a variety of inherited retinal conditions which we hope may reveal new approaches to preserving useful vision for longer.
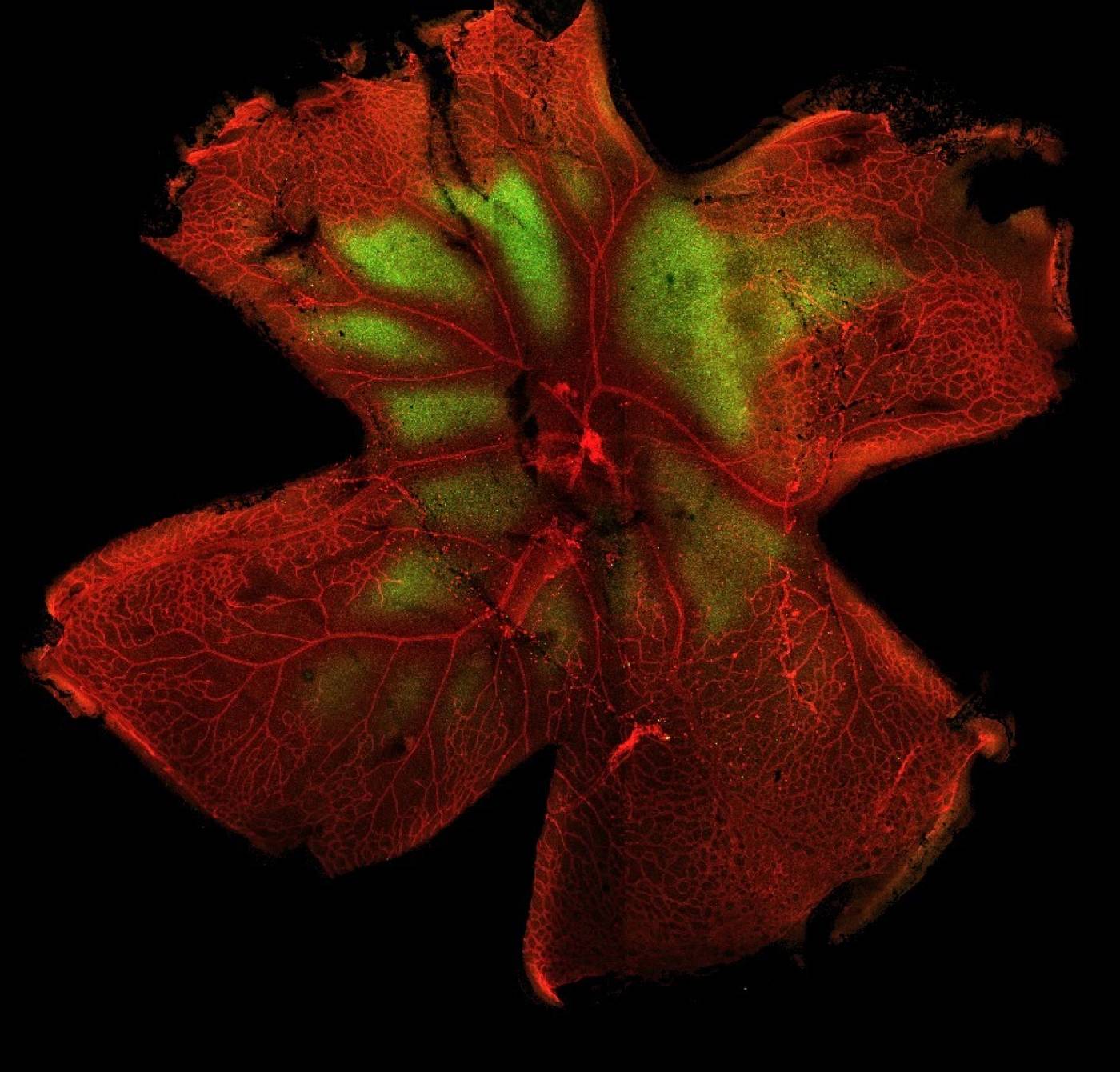
Vascularisation, inflammation and immunity
Because of the high energy demand of the retina, a good blood supply is essential. Consequently, failure of the retinal vasculature to form properly means the retina cannot function normally resulting in degeneration and vision-loss. This can occur in early development such as through the exposure to too high or low levels of oxygen. The vasculature can also be affected in later life such as with conditions like wet age-related macular degeneration or diabetic eye disease where there is a tendency for the abnormal growth of new weak-walled blood vessels which can leak fluid into the retina damaging the surrounding cells (neovascularization).
We know that the pathway responsible for regulating the vasculature of the eye, which includes Vascular Endothelial Growth Factor (VEGF) and the hypoxia-inducible factors (HIFs) that are activated in response to low oxygen levels, also have a role in inflammation and the immune response. An inappropriate immune response is also known to be a consequence of some eye conditions and can result in further progressive damage of the cells of the eye.
We are investigating this pathway to understand its role more fully in eye disease and how we might better control it to reverse or prevent tissue damage and sight loss.