 Close
Close

Professor Robert Brownstone from the Queen Square Institute of Neurology talks us through his lab’s recent work on the role of the spinal cord in Tor1a dystonia.
What does translation mean to you in your work?
We know so little about the brain that to learn more, it is important to recognise that translation in neuroscience is a two-way street. We need to be sure that what we do in the lab is meaningful to our patients. We learn from our patients. We take their problems to the lab, with the long-term aim of bringing our discoveries back to them. Bedside to bench translation is key.
Why were you interested in researching this area?
Dystonias are the third most common movement disorder after Parkinson's Disease and Essential Tremor, affecting nearly 70,000 people across the UK. There are many different types, but in all cases, people suffer from abnormal patterns of muscle contractions. While in some people, this could be something simple, like writers’ cramp, there are others with generalised dystonias affecting the whole body. These can be terribly disabling disorders.
For the last 25 years, we’ve treated genetic dystonias with deep brain stimulation, which requires surgery to put two electrodes into a person’s brain, attached to a pacemaker-type device in the chest. If we do deep brain stimulation for somebody with tremor or Parkinson's Disease, we can tell immediately whether it will be successful. The tremor goes away, or people show signs immediately that the stimulation is helping them.
But with dystonia, there’s no immediate effect: typically, six or eight weeks later, people start to get better. This long delay from treatment to response has always bothered me and that’s how my interest in dystonia started. If we could figure out why there was a delay, we could come up with even better treatments.
What did you set out to learn?
I started to look into dystonia and felt that there was an error in logic in the field. Everything was focused on the basal ganglia – the deep nuclei in the brain.
The logic is that the basal ganglia are important for movement, and diseases of the basal ganglia lead to movement disorders. And then there’s a leap: dystonia is a movement disorder, so it must be a disease of the basal ganglia.
It doesn’t follow. If the disease was in the basal ganglia, why would putting deep stimulating electrodes directly in this area take so long to have an effect? And why could nobody find anything wrong with the basal ganglia in people who had died with the disease? We therefore asked ourselves if the disease could actually be somewhere else.
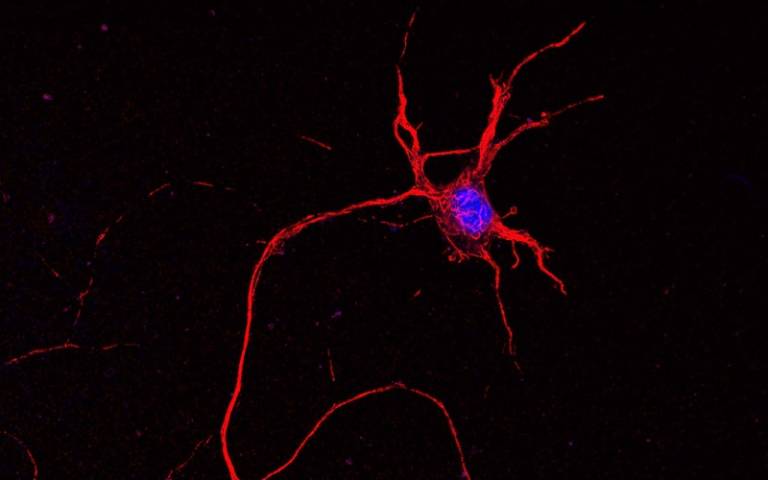
Mouse spinal cord neuron. Nucleus in blue. Credit NICHDS. Jeong
What did your research involve?
We went back to the first principles of neuroscience – to talk to and examine your patients, find out what the deficit is, and based on knowledge of the nervous system, look at where that problem might be. For example, if someone is paralysed from the waist down, their problem is probably in the spinal cord. Dystonias are a disorganisation of the coordination of different muscle contractions, so we asked the crazy question: could these genetic dystonias, in fact, result from disease in the spinal cord, the part of the nervous system responsible for organising muscle contractions?
We took the gene that's involved in the most common form of human dystonia - Tor 1A – and deleted it in the spinal cord only in mice, leaving its expression normal in the brain and rest of the body.
We ended up making a remarkably good model – we were surprised at how similar the movement dysfunction in the mouse model was to that of a human child with DYT-TOR1A dystonia. After validating the model, we studied the circuits of nerve cells in the spinal cord.
When we first presented our preliminary data, somebody said ‘it's just so obvious’, To us, it is obvious, but nobody's ever really looked at it before.
What happens now?
Now that we have identified these circuits in the mouse, we hope to determine whether these same circuits are dysfunctional in humans. We are planning to work with a group in Copenhagen, alongside collaborators in Montpellier, Kings College, and Tel Aviv, to find people with this particular genetic form of dystonia to understand what happens over time to these circuits with deep brain stimulation, which is still our best treatment right now.
When a child is diagnosed, we will fly to their centre and use a range of techniques to study their reflexes, and then six or eight months later, after surgery, we'll go back and see them again. If we find that the disease is in the spinal cord, can we come up with better treatments for dystonia that may be less invasive and more effective than deep brain stimulation?
How will your research extend to other forms of dystonia?
We’re working on the most common form of the genetic dystonias, but even so it is considered an orphan disease. We’ll see what we can learn about movement and movement disorders from studying this particular one before focussing on other forms. But we've already started to look at different mouse models and other genetic forms in the lab.
How has working at Queen Square enabled you to work on translational research like this?
I would never have done this work if I had not come to UCL and been in this rich environment – clinically and scientifically. I can just run into somebody in Queen Square, have a casual conversation and have their input immediately. And for our trainees, it's absolutely essential. Amanda Pocratsky, supported by EMBO and the Dystonia Medical Research Foundation who has been leading on this work, came here as a post doc with no clinical background, but has thrived in part because of this rich environment. She can collaborate with clinical academics and to clinics with dystonia patients so she can see these patients themselves. That’s not going to happen in many places.