 Close
Close

Research
We use NMR spectroscopy together with biochemical and biophysical methods to study these co-translational folding processes with atomic resolution, in order to understand the fundamental principles of folding and misfolding within the cellular environment.
These are our primary areas of research:
- Co-translational protein folding
- Co--translational misfolding
- Studying ribosomes and nascent chains by NMR spectroscopy
- In-cell structural biology
- Cryo-Electron Microscopy
- Ribosome engineering
- Molecular dynamics simulations
- TDP-43
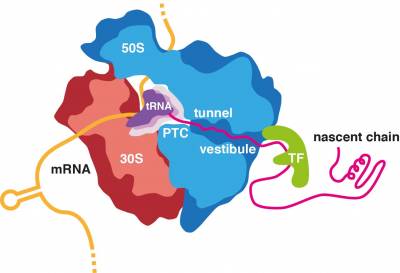
All proteins in all kingdoms of life are synthesized by the ribosome. Most proteins can start to fold during biosynthesis. Moreover, some of the initial misfolding steps can occur while the nascent chain is still attached to its parent ribosome. We are interested in defining the structural and mechanistic principles that underline the fundamental process of co-translational protein folding and, when this process goes askew, identifying the molecular mechanisms of co-translational protein misfolding and aggregation and their links to disease.
We are investigating the essential role of the ribosome in the regulation of folding processes. The ribosome has been shown to increase the efficiency of protein folding and help proteins attain their native structure, but it can also interact with the emerging nascent chain in a manner than delays successful folding, therefore sequestering hydrophobic segments until later residues have been synthesized, delaying folding ahead of co-translational assembly or preventing engagement with downstream chaperones. Our ongoing research is identifying the remarkable extent to which the ribosome can modulate protein folding through sequence specific interactions with the nascent chain both within the ribosome exit tunnel and with the ribosomal surface upon emergence into the crowded cellular environment.
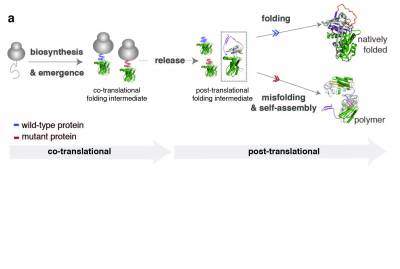
Efficient protein biosynthesis and co-translational folding are essential for maintaining proteostasis within cells, but these processes are often complicated by the competing effects of protein misfolding and aggregation. After release from the ribosome and even while synthesized, the nascent chain faces a choice between native folding and misfolding. Unfolded or partially folded intermediates, are particularly vulnerable to aggregation; under cellular stress (changes in pH, temperature or genetic mutations) they can associate and lead to the formation of protein aggregates. These aggregates accumulate in the cell and result in the formation of toxic inclusions, linked to the onset of over 50 different human diseases collectively referred to as “misfolding diseases’’. Although there is evidence of an early onset for this process, molecular descriptions of co-translational misfolding remain sparse.
In our lab, we provide molecular descriptions of the earliest stages of protein misfolding by monitoring biosynthesis and folding of nascent polypeptide chains and identifying the co-translational intermediates that, upon release from the ribosome, misfold, aggregate and lead to disease. We use biochemical methods like PEGylation and structural methods like NMR spectroscopy and cryo-EM to characterise the ribosome bound nascent chains and investigate the mechanism that leads to misfolding. Better understanding of co-translational misfolding is essential in the design of therapeutic strategies that target the early onset of misfolding diseases. Some of the protein systems we use to study co-translational misfolding are; a1-antitrypsin- a misfolding-prone protein which self-associates and polymerises, causing liver cirrhosis and emphysema within individuals suffering from alpha-1-antitrypsin deficiency- and huntingtin- a protein that contains expansions of glutamines that make it prone to misfolding and aggregation leading to Huntington’s disease.
Further reading
Plessa, E., Chu, L. P., Chan, S. H., Thomas, O. L., Cassaignau, A. M., Waudby, C. A., ... & Cabrita, L. D. (2021). Nascent chains can form co-translational folding intermediates that promote post-translational folding outcomes in a disease-causing protein. Nature communications, 12(1), 1-13.
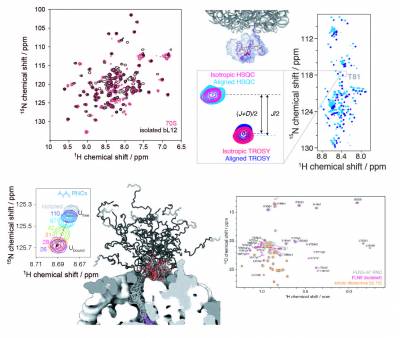
Cryo-electron microscopy and X-ray crystallography studies are providing highly detailed structures of the ribosome, but its more dynamic regions are often less well-resolved and their study therefore requires complementary techniques. We have pioneered the use of solution-state nuclear magnetic resonance (NMR) spectroscopy to examine ribosomal particles, enabling the study of their flexible regions at a residue-specific level. These include the bL12 stalk region of the 70S E. coli ribosome, involved in the recruitment of several auxiliary factors associated with protein synthesis, and the nascent polypeptide chain, which emerges from the ribosome exit tunnel during translation and begins to adopt tertiary structure. Our in vitro and in-cell NMR studies are providing key mechanistic insights into how the ribosome can influence and regulate the co-translational folding pathways, structures, interactions, and thermodynamics of nascent chains during biosynthesis.
The significant technical challenges of NMR studies of ribosomal complexes, arising from low sensitivity due to slow molecular tumbling and limited sample concentrations, combined with their short lifetimes, necessitates development and improvements in biochemical and NMR methodologies. We have established robust protocols to produce high yields of stable, homogenous ribosomal complexes in vivo, selectively labelling the nascent chain with a variety of different isotopes, including 15N, 13C (uniform and methyl-selective with 2H), and 19F, with each providing spectroscopic probes of specific structural conformations of the nascent chain. We also develop NMR methods and tools to enhance spectroscopic sensitivity and to enable the acquisition of quantitative measurements to characterise the structure and dynamics of ribosome-bound nascent chains at high resolution.
Selected publications
Cassaignau, A. M. et al. A strategy for co-translational folding studies of ribosome-bound nascent chain complexes using NMR spectroscopy. Nat Protoc 11, 1492-1507, doi:10.1038/nprot.2016.101 (2016).
Cabrita, L. D. et al. A structural ensemble of a ribosome-nascent chain complex during cotranslational protein folding. Nat Struct Mol Biol 23, 278-285, doi:10.1038/nsmb.3182 (2016).
Cassaignau, A. M. E. et al. Interactions between nascent proteins and the ribosome surface inhibit co-translational folding. Nature Chemistry, doi:10.1038/s41557-021-00796-x (2021).
Deckert, A. et al. Common sequence motifs of nascent chains engage the ribosome surface and trigger factor. Proc Natl Acad Sci U S A 118, doi:10.1073/pnas.2103015118 (2021).
Wang, X. et al. Probing the dynamic stalk region of the ribosome using solution NMR. Sci Rep 9, 13528, doi:10.1038/s41598-019-49190-1 (2019).
Burridge, C. et al. Nascent chain dynamics and ribosome interactions within folded ribosome–nascent chain complexes observed by NMR spectroscopy. Chemical Science, doi:10.1039/d1sc04313g (2021).
Chan, S. H. S., Waudby, C. A. & Christodoulou, J. NMR snapshots of nascent chains emerging from the ribosome during biosynthesis. ChemRxiv, doi:10.26434/chemrxiv-2022-0lmsp (2022).
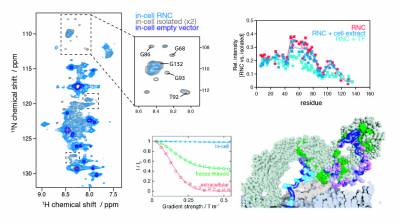
As the newly synthesised nascent chain emerges from the ribosome exit tunnel and into the crowded cellular environment, a variety of co-translational processes can occur alongside folding. The ribosome acts as a central hub to such processes, regulating quality control to maintain protein homeostasis, which involve ribosome-associated molecular chaperones such as trigger factor and Hsp70, and the translocation, targeting and modification of nascent proteins. To study these cellular processes, we employ and develop methods in in-cell NMR spectroscopy and electron tomography. Our recent findings include the first observations of ribosome-nascent chain complexes in live bacterial cells and uncovering the sequence determinants of trigger factor interactions with the nascent chain.
Selected publications
Deckert, A. et al. Common sequence motifs of nascent chains engage the ribosome surface and trigger factor. Proc Natl Acad Sci U S A 118, doi:10.1073/pnas.2103015118 (2021).
Waudby, C. A. et al. Rapid distinction of intracellular and extracellular proteins using NMR diffusion measurements. J Am Chem Soc 134, 11312-11315, doi:10.1021/ja304912c (2012).
Waudby, C. A. et al. In-cell NMR characterization of the secondary structure populations of a disordered conformation of alpha-synuclein within E. coli cells. PLoS One 8, e72286, doi:10.1371/journal.pone.0072286 (2013).
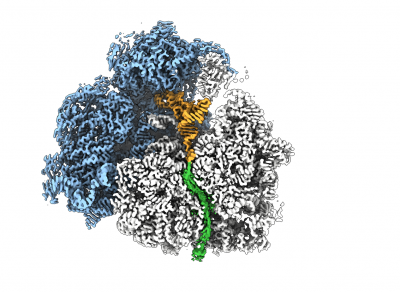
To study the nascent chain (NC) within the tunnel as it emerges from the ribosomal exit tunnel, we combine NMR spectroscopy and MD simulations with state-of-the-art cryo electron microscopy (cryoEM). CryoEM is a technique that enables us to visualise what is in a biological sample by vitrifying it to preserve its state and acquire images directly without prior crystallisation or other time- and sample-consuming processes. The use of special transmission electron microscopes and new direct detection cameras that use real-time electron counting allow for fast and super resolution data acquisition. These images (large datasets) are then being processed with single particle analysis using a wide range of software and high-speed processing GPU and CPU cores for data processing and analyses.
The combination of cryoEM with the biochemical and structural techniques we use in the lab enables us to build atomic models of the RNC to visualise co-translational protein folding at high resolution and study the influence of the ribosome on this process. The resulting maps show that we are able to visualise a 3D ensemble of folded protein, and by deconvoluting each state, we find that each occupies a distinct binding site on the ribosomal surface near the exit tunnel.
We have access to the ISMB Electron Microscopy Facility (Birkbeck College, University of London) and to Cryo-EM facilities at the UK national electron bio-imaging centre (eBIC) at Diamond.
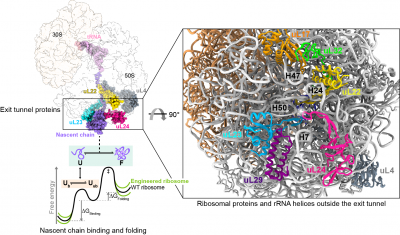
Protein folding begins during biosynthesis on ribosomes as nascent polypeptide chains exit via the ribosome’s narrow exit tunnel. This universal conduit is crafted from ribosomal RNA and uniquely shaped by a series of evolutionarily-conserved ribosomal proteins that usher the fledgling nascent chain into the cellular environment. Also, the surface of the ribosome outside the exit tunnel is important for ribosome-nascent chain interactions and recruiting ribosome-associated factors such as molecular chaperones that help assist co-translational folding.
To understand how the ribosomal proteins and RNA modulate co-translational folding and to explore the potential to utilise these mechanisms, we have exploited structure-led rational design and engineered the ribosome by CRISPR/Cas9 gene editing technology, specifically remodelling the ribosomal exit tunnel and outside surface. Using an integrative structural biology toolkit which merges solution NMR spectroscopy, single-particle cryo-EM, and all-atom molecular dynamics simulations we study how an engineered ribosome alters the free energy landscape of co-translational protein folding. The ability to manipulate nascent chain folding outcomes without altering their genetic codes provides useful principles for how to remodel the ribosome to elicit a desired folding outcome.
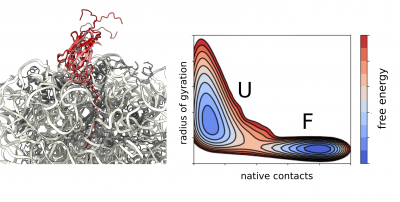
We apply molecular dynamics simulations as part of our integrative structural biology strategy to obtain high-resolution structural ensembles of ribosome-bound nascent polypeptides. We use experimental data from NMR and cryo-EM as structural restraints to guide our simulations directly or we apply them a posteriori during reweighting of the generated structural ensemble.
Using state-of-the-art all-atom and coarse-grained force fields we study the dynamics and structural properties of ribosomal nascent chains, which are complex molecular systems that are typically composed of millions of atoms. These simulations combined with enhanced sampling methods such as metadynamics have already been very accurate in predicting the onset of co-translational folding, ribosome-nascent chain interactions that can modulate folding, as well as the structure of folding intermediates. In combination with experimental data, these simulations are providing the first insights into co-translational folding at atomic resolution. Moreover, we increasingly employ observations obtained through simulations to directly guide our future experimental work.
We have access to the following facilities:
Archer2 (Tier 1, UK)
Okeanos (Interdisciplinary Centre for Mathematical and Computational Modelling University of Warsaw, Poland)
Baskerville (Tier 2, UK)
Young (MMM Hub, Tier 2, UK)
UCL Research Computing Platforms (Tier 3, UK)
Further reading:
1 Cassaignau, A. M. E. et al. Interactions between nascent proteins and the ribosome surface inhibit co-translational folding. Nature Chemistry, doi:10.1038/s41557-021-00796-x (2021).
2 Deckert, A. et al. Common sequence motifs of nascent chains engage the ribosome surface and trigger factor. Proc Natl Acad Sci U S A 118, doi:10.1073/pnas.2103015118 (2021).
3 Cabrita, L. D. et al. A structural ensemble of a ribosome-nascent chain complex during cotranslational protein folding. Nat Struct Mol Biol 23, 278-285, doi:10.1038/nsmb.3182 (2016).
Motor neurone disease/Amyotrophic lateral sclerosis (MND/ALS) involves the progressive and premature loss of motor neurons within the central nervous system, which leads to muscle weakness, atrophy and ultimately fatal paralysis. Within the motor neurons of MND/ALS patients are intracellular aggregates of a protein, trans-activation response DNA binding protein 43 (TDP-43). TDP-43 is a DNA/RNA binding protein whose function includes roles in transcription, mRNA splicing, and translational regulation. This large 43kDa multi-domain protein consists of an N-terminal nuclear localisation domain (NTD), followed by two highly conserved RNA-binding motifs (RRM1-RRM2), and a highly disordered and aggregation-prone glycine-rich C-terminal domain (CTD274-414).

Currently very little is understood of TDP-43's overall 3D structure and its role in function, and how this protein reorganises its architecture so that it misfolds and forms aggregates. Using a combination NMR spectroscopy, computational approaches and biophysics, we are investigating the structural and dynamical properties of this fascinating protein and its conformational preferences within the cell.
Since the 1960s, 1D lineshape fitting has been a key method for the quantitative analysis of titration data. Today however, even applications of 1D lineshape analysis have become limited due to the spectral complexity associated with increasingly large biomolecules.
We've developed a simple and practical extension of lineshape fitting to 2D spectra that can resolve overlapping peaks and eliminate systematic errors, bringing improved accuracy and convenience.

We have developed TITAN, an easy-to-use software package for the analysis of NMR titrations in two dimensions:
- Import data from nmrPipe
- Fit 2D data to a variety of binding models
- Create overlays of fitted spectra with a single click
- Use bootstrap methods to calculate rigorous error estimates
TITAN is free for academic users! Please register here to download (link will open in new window), or (registered users) access the source code repository.
Two-Dimensional NMR Lineshape Analysis Paper