Close
Close

Neuroscience, Physiology and Pharmacology
Welcome
From founding the first department of Physiology in England in 1828 to its groundbreaking work in Pharmacology to its current position as part of the largest research community of Neuroscientists in Europe, this research department has been instrumental in the world's understanding of the nervous system and the interactions within it.
From James Black to Archibald Hill to Bernard Katz, UCL has been home to Nobel Prize winners and game changers in the discipline of physiology.
Today NPP continues this fine tradition with some of the most prestigious academics in the field. From Maria Fitzgerald and her groundbreaking work on pain to David Attwell's important work on the control of blood flow to Annette Dolphin's work on how neurons talk to each other and many more, UCL's many NPP research labs are generating exciting new findings daily.
We are proud of our focus on research-based teaching and offering the top neuroscientists of tomorrow the opportunity to hear from the current leaders in the field on their areas of expertise as well as ensuring that practical experience in labs remains a fundamental element of the teaching process.
Stephanie Schorge - Head of Department
Our people and our history



Research
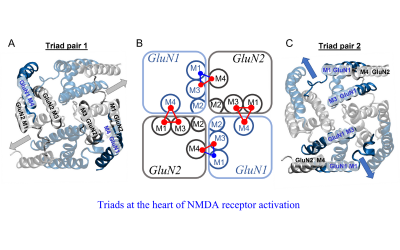
Molecular and Cellular Neuroscience
Our work focuses on the study of ligand- and voltage-gated ion channels, G-protein-coupled receptors, and organelles, from structural and functional perspectives to their role in synaptic transmission during health and disease.
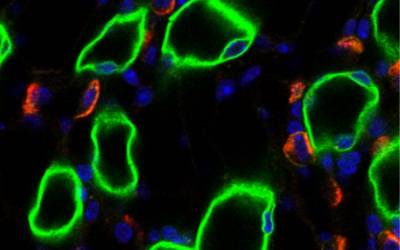
Epithelial Physiology
Our work focuses on the investigation of the function of channels and transporters in epithelial membranes, in relation to homeostasis and disease.
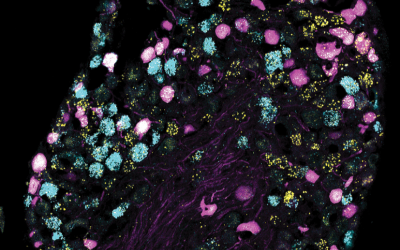
Circuits and Systems Neuroscience
Research in the Circuits and Systems laboratories in the department focuses on understanding the fundamental mechanisms of circuit and system function and how it contributes to emerging behaviours in health and disease
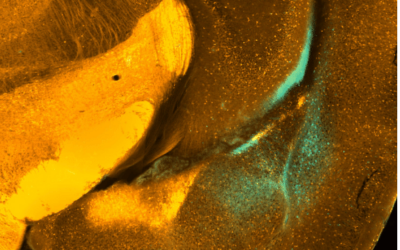
Our research spans receptor pharmacology, biophysics, circuits and systems neuroscience, cardiovascular neurophysiology and neural computation. We encourage early-career scientists to join us with externally funded fellowships.
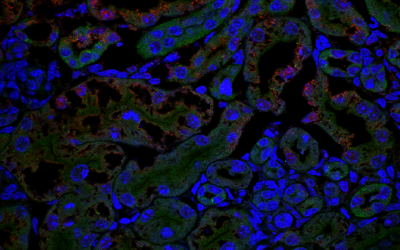
Early Careers Community
Our research department is home to a thriving community of Postdoctoral Researchers supported by skills training, careers advice and much more.

Our staff, researchers and students have access to a wide range of cutting-edge facilities within NPP and Biosciences as well as across UCL.
Study

Undergraduate
UCL offers two of the most prestigious undergraduate programmes in this field in the world: BSc/MSci Neuroscience and BSc/MSci Pharmacology. We also have one of the largest student-led science-focused societies in the UK, the UCL Neuroscience Society.

Masters
We offer a dedicated MSc in Neuroscience and we have streams in both Neuroscience and Pharmacology in our MRes Biosciences.
PhD
Prospective students are warmly encouraged to review the many research topics in the department and to make early contact with academic staff. PhD projects may be based entirely within NPP, or connected to collaborative research projects that link departments across UCL and with external partners.
UCL Neuroscience Symposium
Discover some of the most exciting research taking place in neuroscience today as UCL researchers, from established senior investigators to rising stars, present their more recent work. This immensely popular forum has been running for 12 years and attracts over 800 delegates each year.