 Close
Close

Graham S Jackson
 g.s.jackson@prion.ucl.ac.uk Tel: 020 7679 5028 Courtauld Building, Room 101A • UCL Profile • PubMed • |
Research Synopsis
Prion-like mechanisms have been proposed as central to the spread of infection and worsening of symptoms in a range of neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease. These proposals have been based upon the discovery of prions as the infectious agent of transmissible spongiform encephalopathies (TSEs) such as bovine spongiform encephalopathy (BSE), Creutzfeldt Jakob disease (CJD) and sheep scrapie. Prions are composed of misfolded, ordered aggregates of a host’s normal prion protein (PrP) that are capable of infecting new hosts and carrying phenotypic information that determines disease characteristics. Prions have features in common with simpler amyloid fibrils and indeed may also contain amyloid structure. Both prions and PrP amyloids propagate in parallel within a host following the establishment of a prion infection, but the simpler amyloids are not associated with any infectivity, all of which is attributable to the prions. What distinguishes prions from amyloid is crucial to understanding the difference between prion-like, ie amyloid properties and infectious prions themselves.
A method to robustly replicate prions in vitro was first described in 2001 and was termed protein misfolding by cyclic amplification (PMCA). PMCA typically requires a substrate produced from homogenised brain tissue that contains PrPC which limits the ability to adapt the substrate for study. We have recently developed a modified PMCA reaction that generates high prion titres from recombinant PrP expressed in bacteria. In order to generate infectious prions rather than simply amyloid, the reaction still requires the addition of brain homogenate. However, in these modified reactions the brain homogenate is provided from a transgenic mouse line that does not produce any PrPC. The PrP is provided separately in recombinant form and hence can be manipulated using standard, genetic engineering techniques, facilitating further study.
This represents a quantum leap forward in our ability to understand what differentiates non-infectious PrP amyloids from prions as the high titres generated coupled with recent advances in the ability to purify prions to heterogeneity allow not only manipulation by standard genetic and biochemical techniques, but also the isolation of these synthetic prions for further studies.
We are interested in defining the mechanism of prion replication at the atomic level which includes determining the structures of synthetic prions by cryo-electron microscopy and identifying the essential components contained within a brain homogenate that are required for prion replication in vitro. The crucial requirement for brain homogenate as a supplement to replicate prions rather than amyloid is being investigated, both by fractionation and modification of the brain homogenate substrate and by mass spectrometric analysis of the purified synthetic prions to determine what components additional to PrP are required to generate a prion.
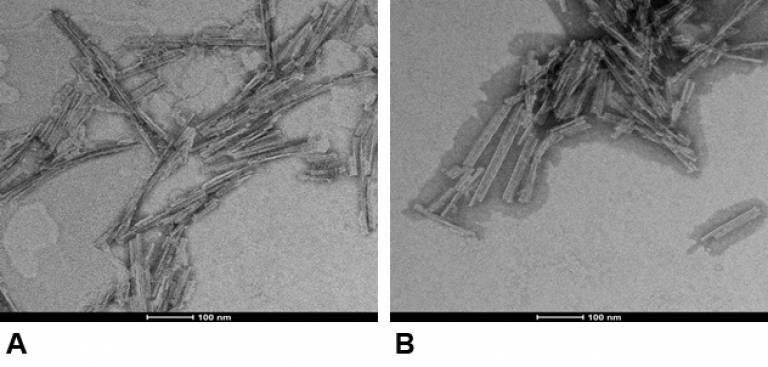 |
Negatively stained electron micrographs of prion rods purified from (A) infected mouse brain, (B) synthetically generated in vitro. |
Selected Publications
Neurofilament light chain and tau concentrations are markedly increased in the serum of patients with sporadic Creutzfeldt-Jakob disease, and tau correlates with rate of disease progression.
Thompson AGB, Luk C, Heslegrave AJ, Zetterberg H, Mead SH, Collinge J, Jackson GS J Neurol Neurosurg Psychiatry. 2018 Feb 27.
Diagnosing Sporadic Creutzfeldt-Jakob Disease by the Detection of Abnormal Prion Protein in Patient Urine.
Luk C, Jones S, Thomas C, Fox NC, Mok TH, Mead S, Collinge J, Jackson GS. JAMA Neurol. 2016 Oct 3. doi: 10.1001/jamaneurol.2016.3733. PubMed PMID: 27699415.
Preclinical detection of infectivity and disease-specific PrP in blood throughout the incubation period of prion disease.
Sawyer EB, Edgeworth JA, Thomas C, Collinge J, Jackson GS. Sci Rep. 2015 Dec 3;5:17742. doi: 10.1038/srep17742.
Population Screening for Variant Creutzfeldt-Jakob Disease Using a Novel Blood Test: Diagnostic Accuracy and Feasibility Study
Jackson G, Burk-Rafel J, Edgeworth J, Sicilia A, Abdilahi S, Korteweg J, Mackey J, Thomas C, Wang G, Schott J, Mummery C, Chinnery P, Mead S, Collinge J. JAMA Neurol 2014; 71: 421-8.
Detection of prion infection in variant Creutzfeldt-Jakob disease: a blood-based assay.
Edgeworth JA, Farmer M, Sicilia A, Tavares P, Beck J, Campbell T, Lowe J, Mead S, Rudge P, Collinge J, Jackson GS. Lancet 2011; 377: 487-93.
Discrimination between prioninfected and normal blood samples by protein misfolding cyclic amplification
Tattum MH, Jones S, Pal S, Collinge J, Jackson GS. Transfusion 2010; 50: 996-1002.