 Close
Close

Lysosomes are cell structures that contain more than 50 different enzymes. These enzymes can break down a range of molecules, including waste from the cell, and even the cell itself when it dies.
Mutations of lysosomes can cause problems with the enzymes and how they work. This can result in a range of conditions that affect whole organ systems, called lysosomal storage diseases.
The Lysosomal Storage Disorders (LSD) Unit at the Royal Free is one of the largest designated specialist units in the UK, currently treating around 400 patients.
Our major focus is on treating adult patients in a large, multi-disciplinary hospital, where a range of specialists have experience in the broad range of symptoms associated with these disorders.
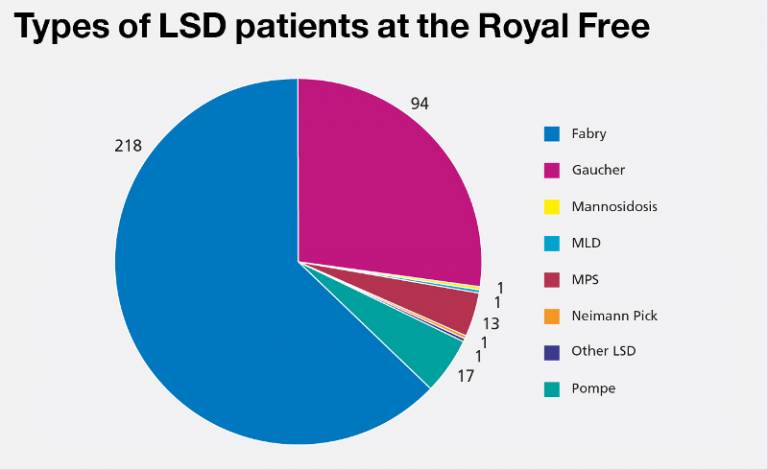
Fabry disease is the most common LSD. It is inherited, and includes more females than males, but males are more severely affected.
Gaucher disease is the second most common LSD. Most of our patients have the adult form of the condition (Type 1), but a growing number have more severe Gaucher disease (Type 3).
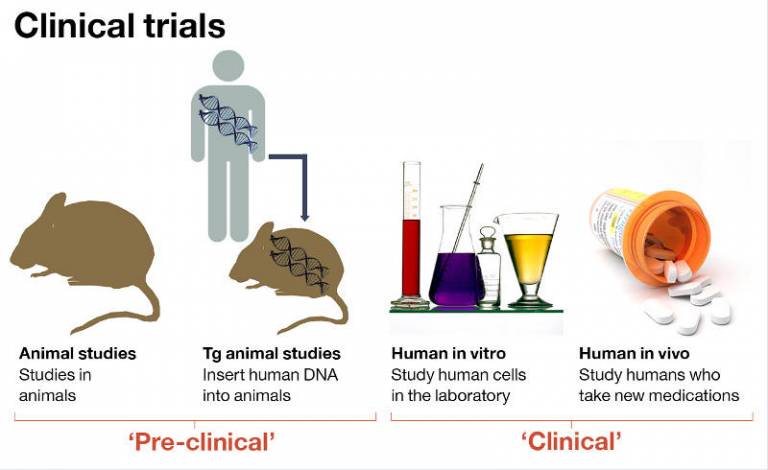
We have been at the forefront of facilitating access to the latest treatments for patients with LSDs, working closely with UCL and our commercial partners.
The unit has led or participated in clinical trials of almost every therapeutic agent approved for lysosomal stroage disorders in the last 10 years.
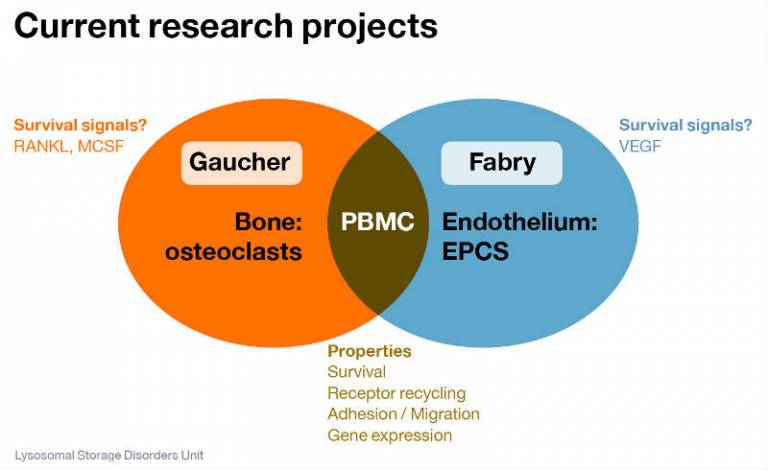
Our major research interests are focused on Anderson-Fabry Disease and Gaucher Disease.
We are trying to improve our understanding of the way in which glycoshingolipid accumulation results in individual patient pathology.

Our clinical services are patient focused, so that patients with rare diseases can access appropriate specialist services provided by professionals who are familiar with their condition.
We liaise closely with local service providers and GPs, operate a 24-hour telephon advisory service and work closely with local specialised teams.