 Close
Close


UNDERSTANDING PRINCIPLES GOVERNING DISEASE SUSCEPTIBILTY AND CLINICAL VARIABILITY IN CILIOPATHIES
Building on human genetic discoveries in skeletal ciliopathies, the Jenkins lab investigates fundamental genetic principles and mechanisms that explain clinical variability in patients carrying mutations in the same gene.
We discovered RAB23 mutations in Carpenter syndrome which, alongside the discovery of IFT80 mutations in Jeune syndrome, defined the skeletal ciliopathies as a new category of human genetic disease caused by mutations in core ciliary trafficking proteins.
Cilia are membrane-encased microtubular projections found on the surface of most cell types within the body. They act as signalling hubs which concentrate signal transduction proteins. All proteins required for cilia formation and function must be transported to the ciliary base, and subsequently many of these proteins are transported up and down the ciliary microtubules through the process of intraflagellar transport (IFT) (Figure 1). These transport processes are necessary for posttranslational modifications of signalling proteins and proper regulation of signal transduction. Many IFT genes are mutated in skeletal ciliopathies demonstrating that limbs, bone, eyes and kidneys are the most susceptible tissues when these core ciliary trafficking processes are disrupted.
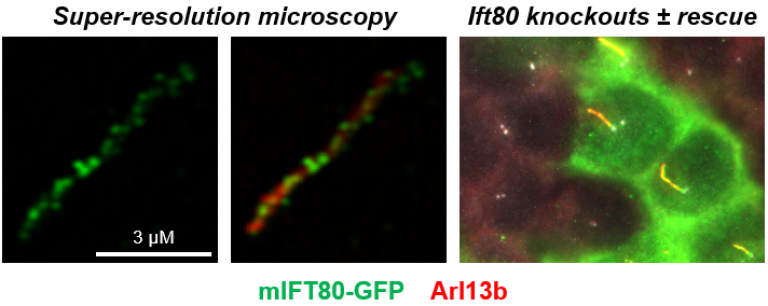
Figure 1. Knockout of Ift80 completely prevents ciliogenesis in IMCD3 cells. The figures show knockout cells rescued following transfection of mIFT80-GFP. Super-resolution microscopy showed that there is an average of 21.8 ± 9.6 (95% CI) particles per cilium separated by 0.33 ± 0.15 µm (95% CI). In the right-hand panel, rescue of ciliogenesis can be seen in transfected cells (green), but adjacent untransfected cells do not form cilia. We have used this assay to demonstrate the requirement for Ift80 in ciliogenesis and to test the functionality of missense mutations in the gene. See Taschner et al. 2018 for details.
Specific questions that we wish to answer are:
- Why do patients with mutations in the same gene present with different disease severities and what are the mechanisms that account for clinical variability?
- How do stochastic processes determine clinical outcomes in complex morphological traits?
- Why are some tissues more susceptible to disease than others?
Generality of our approach:
In tackling these questions for skeletal ciliopathies, we are always thoughtful of developing concepts and experimental approaches that can be applied widely to many other genetic diseases. As such, we use skeletal ciliopathies as a paradigm to study autosomal recessive genetic diseases in general. Most IFT proteins are WD40-domain containing proteins which is one of the largest protein families in humans. Our focus on missense mutations and splicing represents mechanisms that are directly relevant to many syndromes featuring multiple affected tissues.
In relation to the specific questions listed above, our work broadly falls into three overlapping themes:
Theme 1 – Missense mutations and allelic variation.
Theme 2 – Stochastic splicing processes and dynamical systems modelling.
Theme 3 – Whole organism phenotyping in isogenic genetic models of human mutations.
Research summary
A significant subset of autosomal recessive Mendelian diseases (we estimate 10-20%) are disproportionately enriched for missense mutations, and rarely or never carry frameshift or nonsense mutations in causative genes. These missense mutations are therefore likely to be hypomorphic. The aim of this work is to use these hypomorphic missense mutations to dissect protein interaction networks and genetic threshold effects in different tissues, which will allow us to prioritise disease mechanisms for different phenotypes.
We are formally testing these hypotheses through experimental comparison of the effects of frameshift and missense mutations on quantitative protein-protein interactions and cellular and organismal phenotypes. We have shown that different missense mutations can have radically different phenotypic consequences when we have engineered these exact patient mutations in isogenic lines of mice (Figure 2). Together with collaborators in Germany and London, we have shown that for some genes and the proteins they encode, this correlates with the location of each mutation within the protein and the effects on IFT complex formation. In other genes, we have found unexpected roles for missense mutations in inducing alterative splicing, leading to stochastic levels of functional rescue which varies between individuals and tissues.
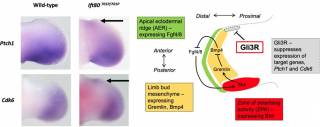
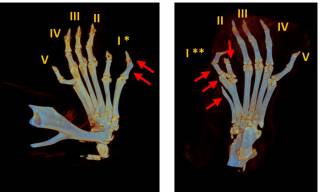
Figure 2. Developing mouse limb buds showing expression of direct target genes of Gli3R, Ptch1 and Cdk6. As outlined in the cartoon to the right, Gli3R is a transcriptional repressor of Ptch1 and Ckd6 which binds directly to the chromatin of these genes and forms an essential part of a dynamic signalling relay essential for limb bud development. Ift80701P/701P is an exact genetic mouse model of Jeune syndrome carrying an p.A701P missense mutation in the Ift80 gene which is found in patients with the disease. These mice show ectopic expression of Gli3R target genes in the anterior limb bud (arrows).Below, microCT scanning shows preaxial polydactyly that results from these signalling defects in Ift80701P/701P mice. The arrows show extra digits – note the variability between genetically identical mice.
Project partners
Phil Beales, Marius Ueffing, Karsten Boldt, Rob Russell, Damian Smedley, Fiona Watt.
Collaborators
Esben Lorentzen, Anthony Roberts.
