 Close
Close

Cell and Developmental Biology
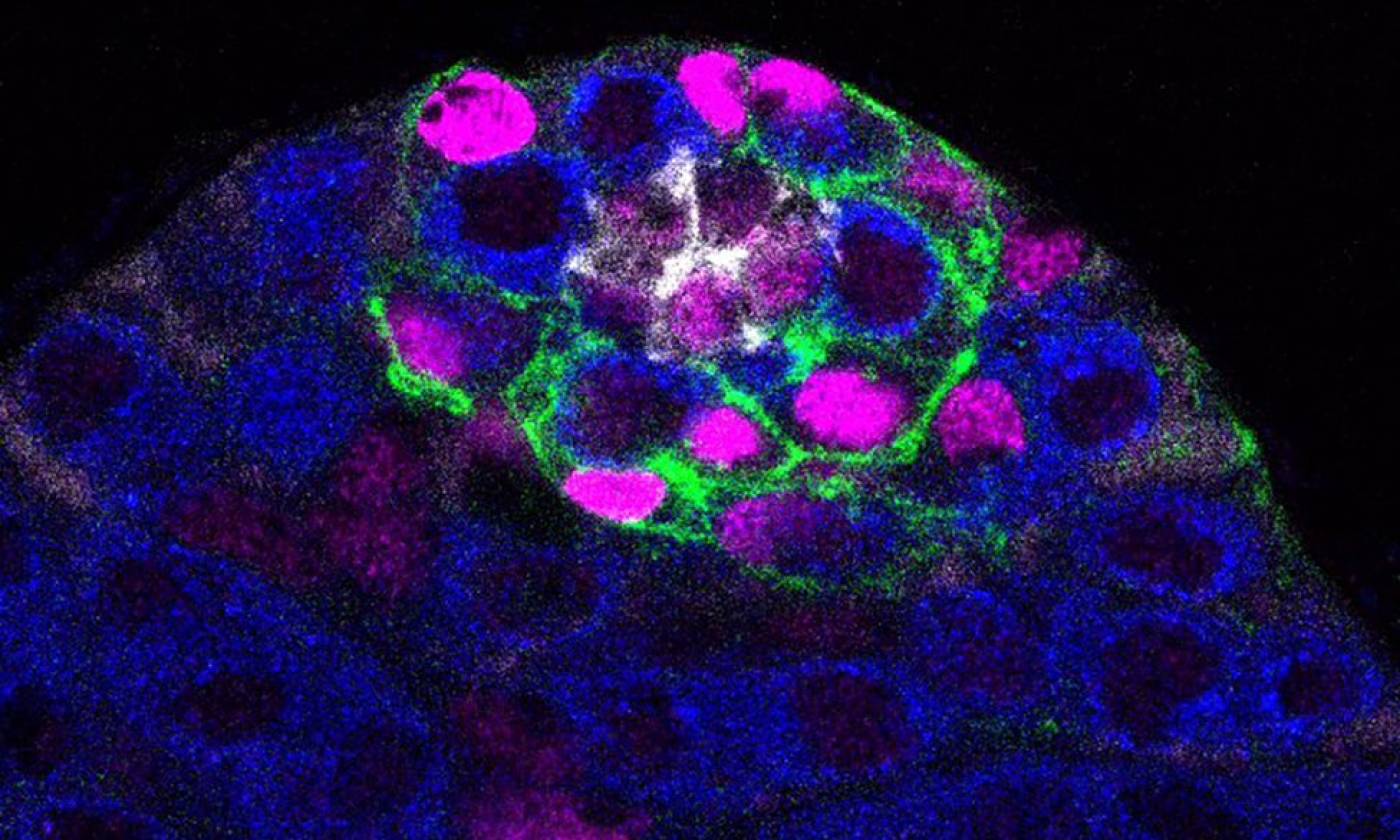
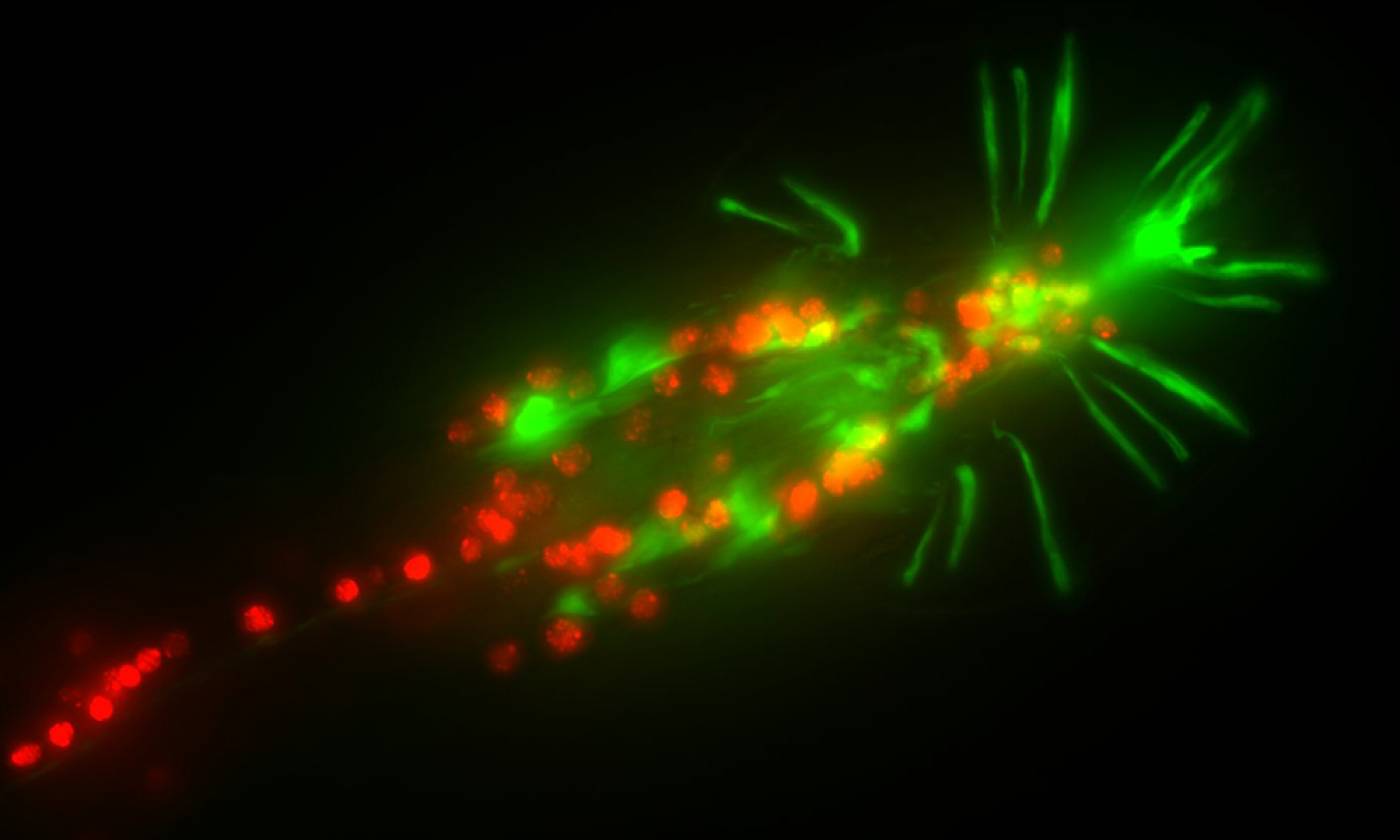
Work in the Barrios lab is focused on genes, circuits and behaviour
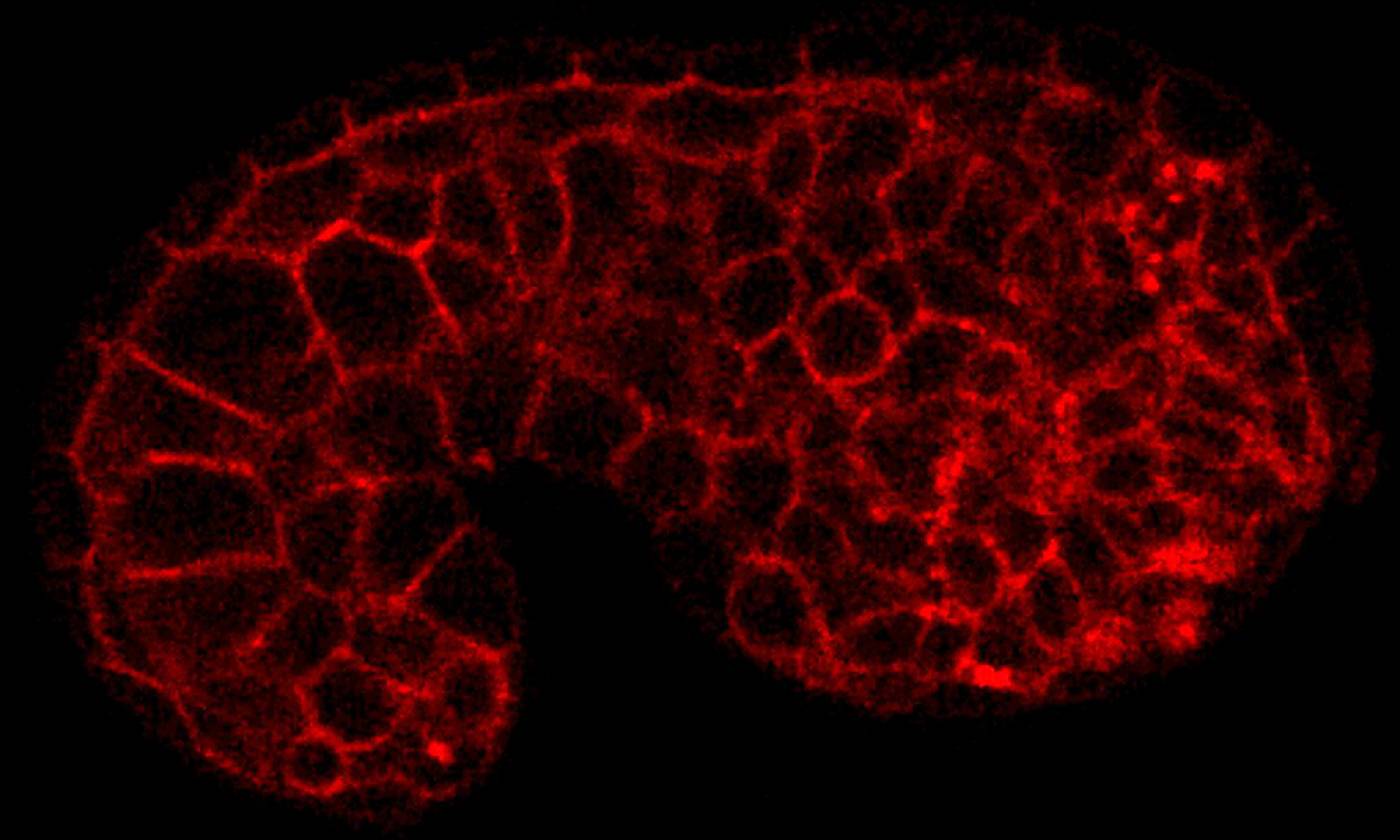
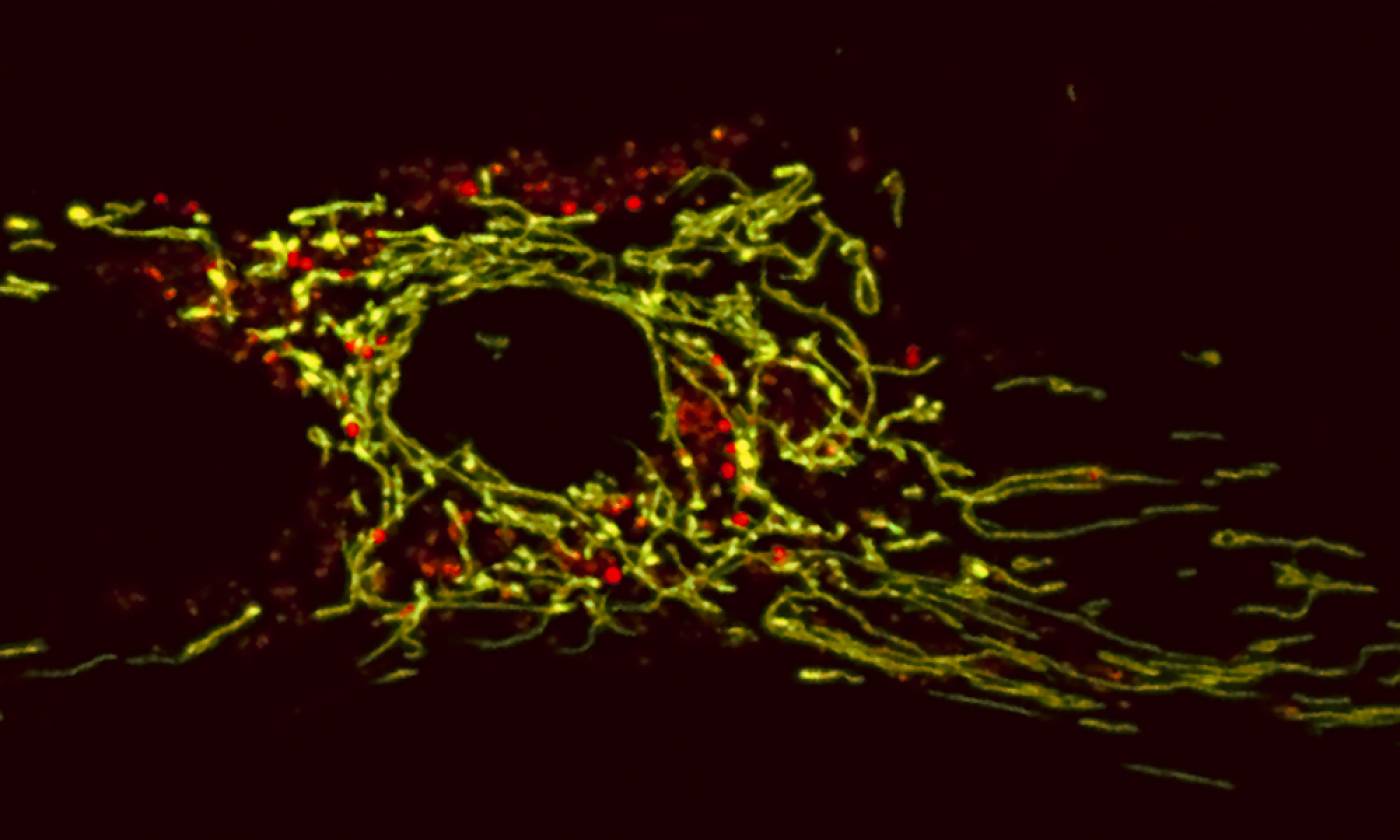
The Duchen lab studies the regulation of energy homeostasis in health and disease
The Evans lab studies the evolutionary history of reptiles and amphibians
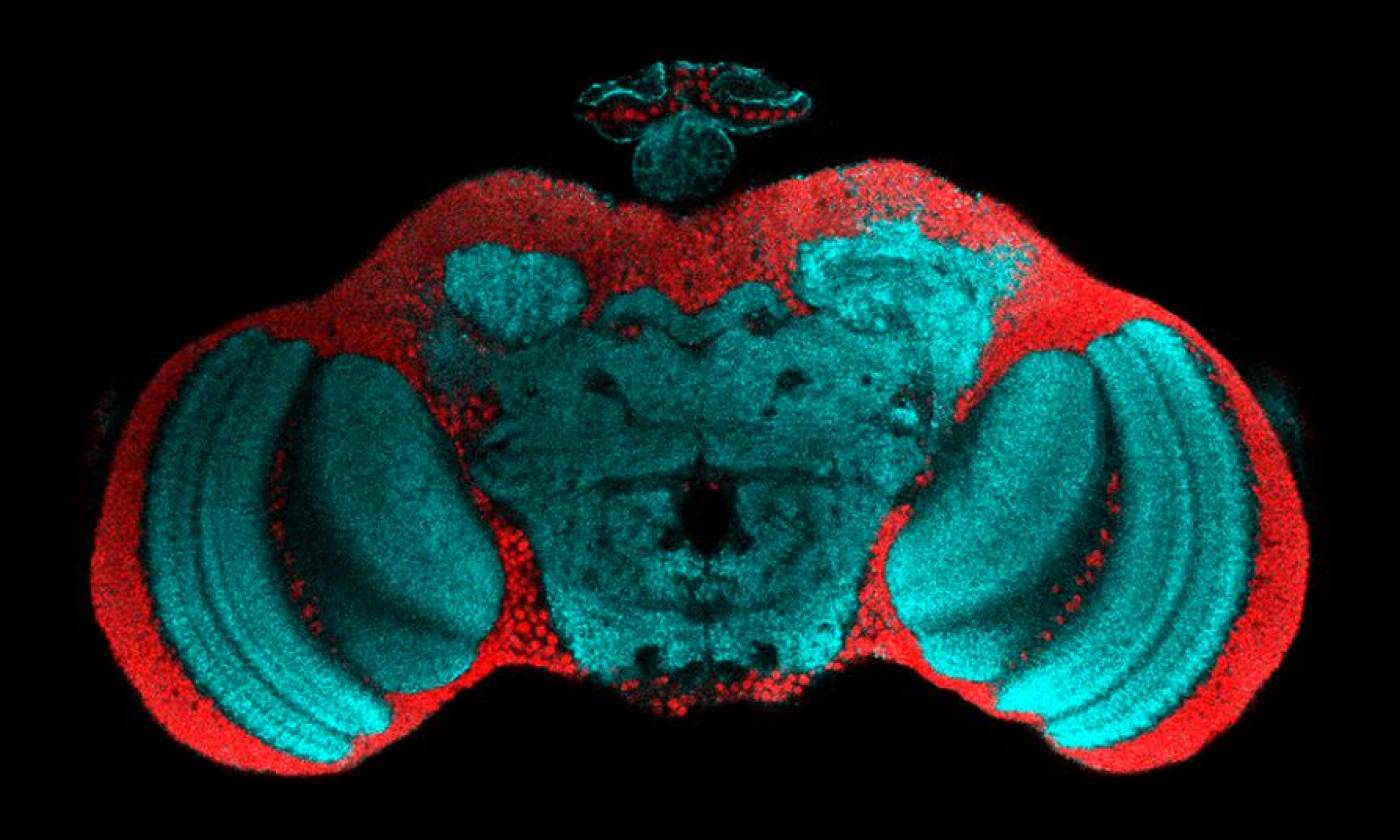
The Fernandes lab studies glia-neuron interactions during development
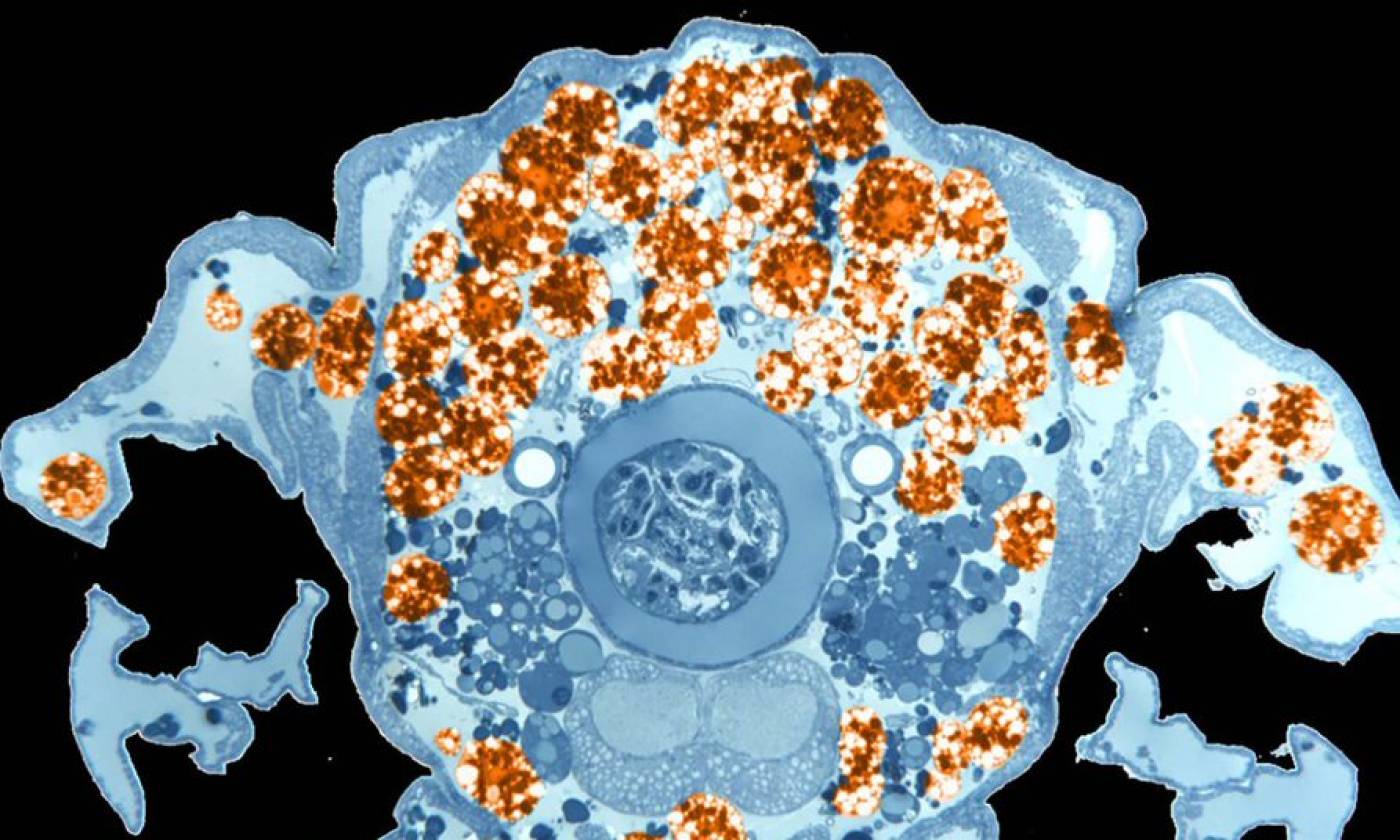
The Franz lab expores the roles of motile adipocytes in wound healing and cancer
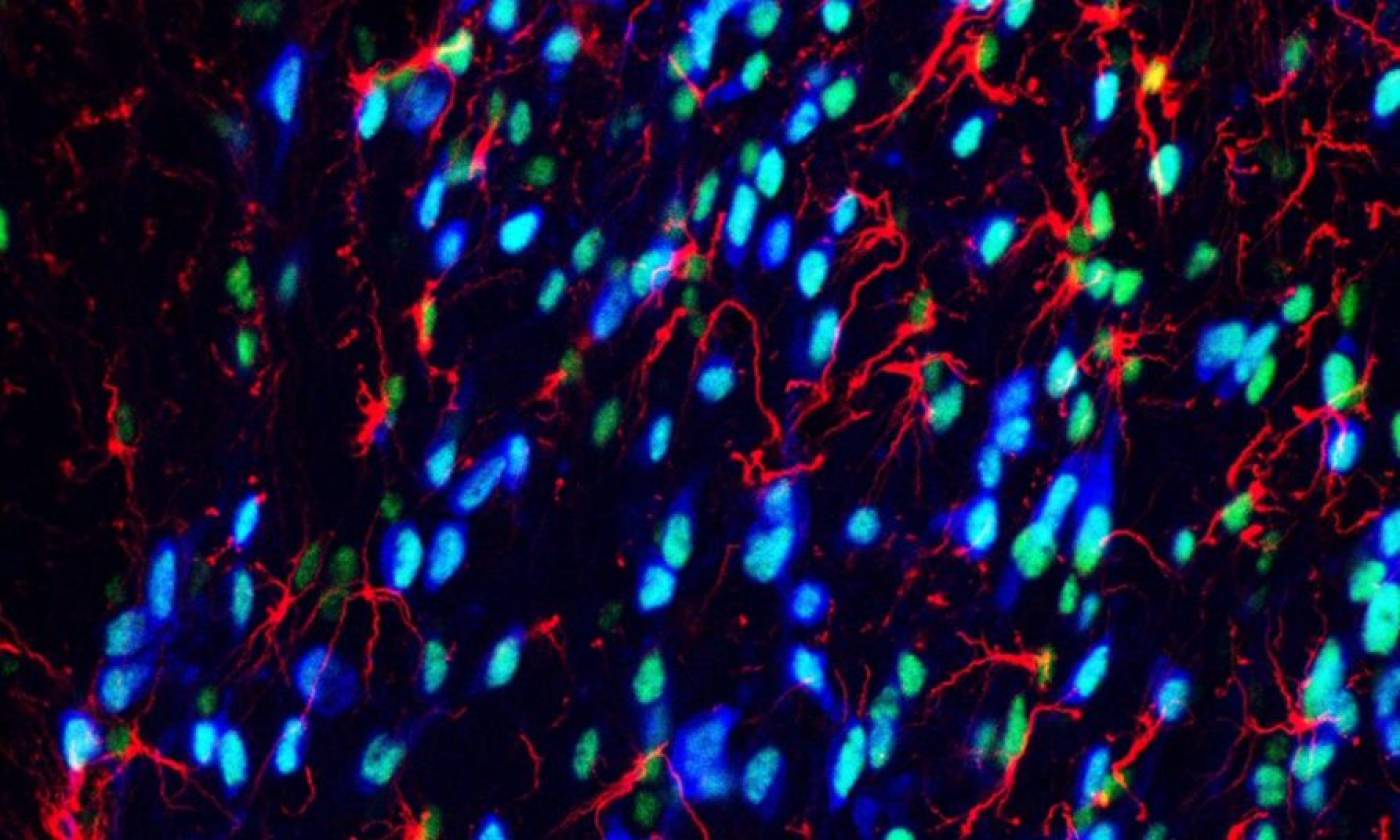
Work in the Geranton lab is focused on the molecular biology of pain
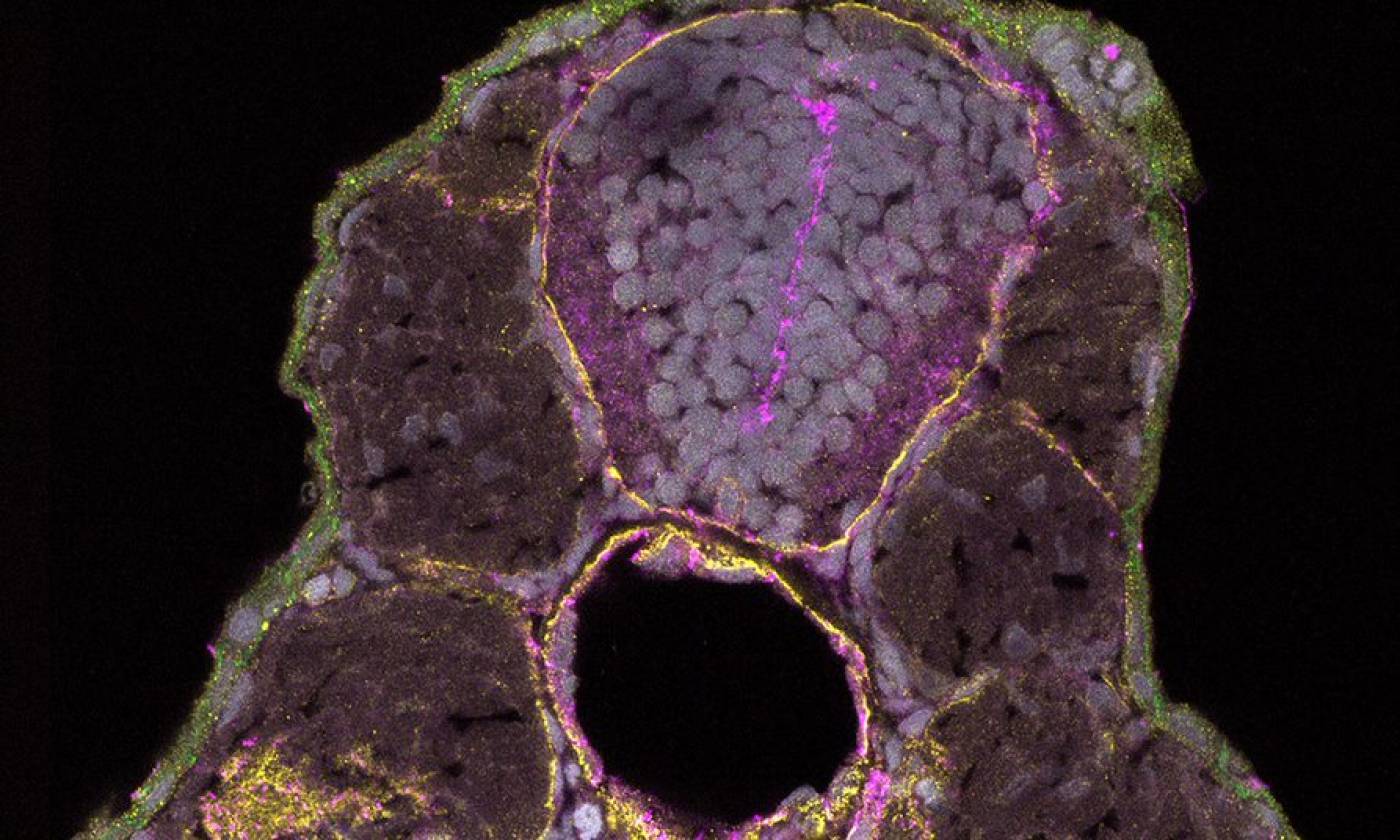
The Hawkins lab studies neuroanatomy and development using the zebrafish
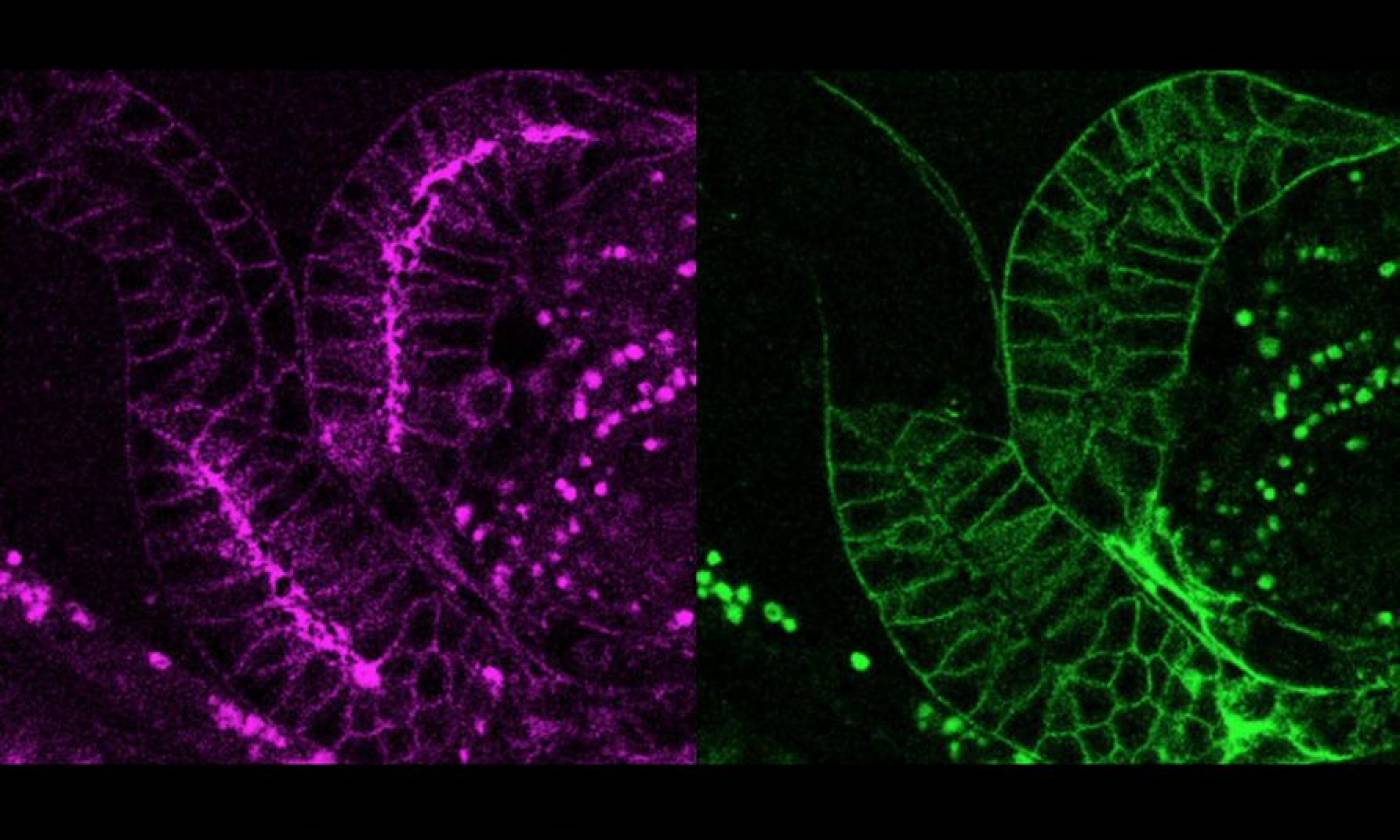
The Lambie Lab studies the function and regulation of P-type ATPases
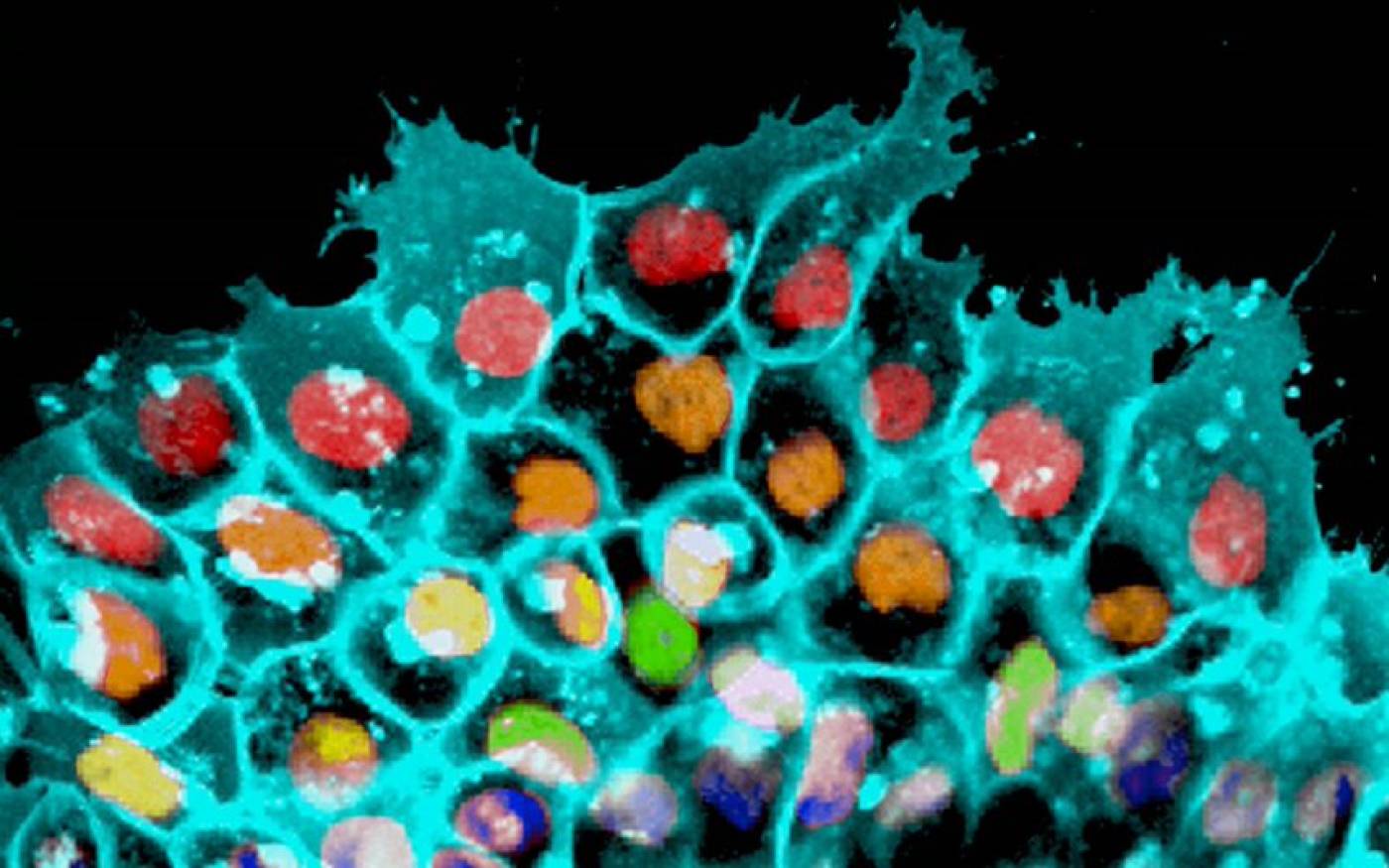
The Mayor lab studies cell migration and differentiation during development
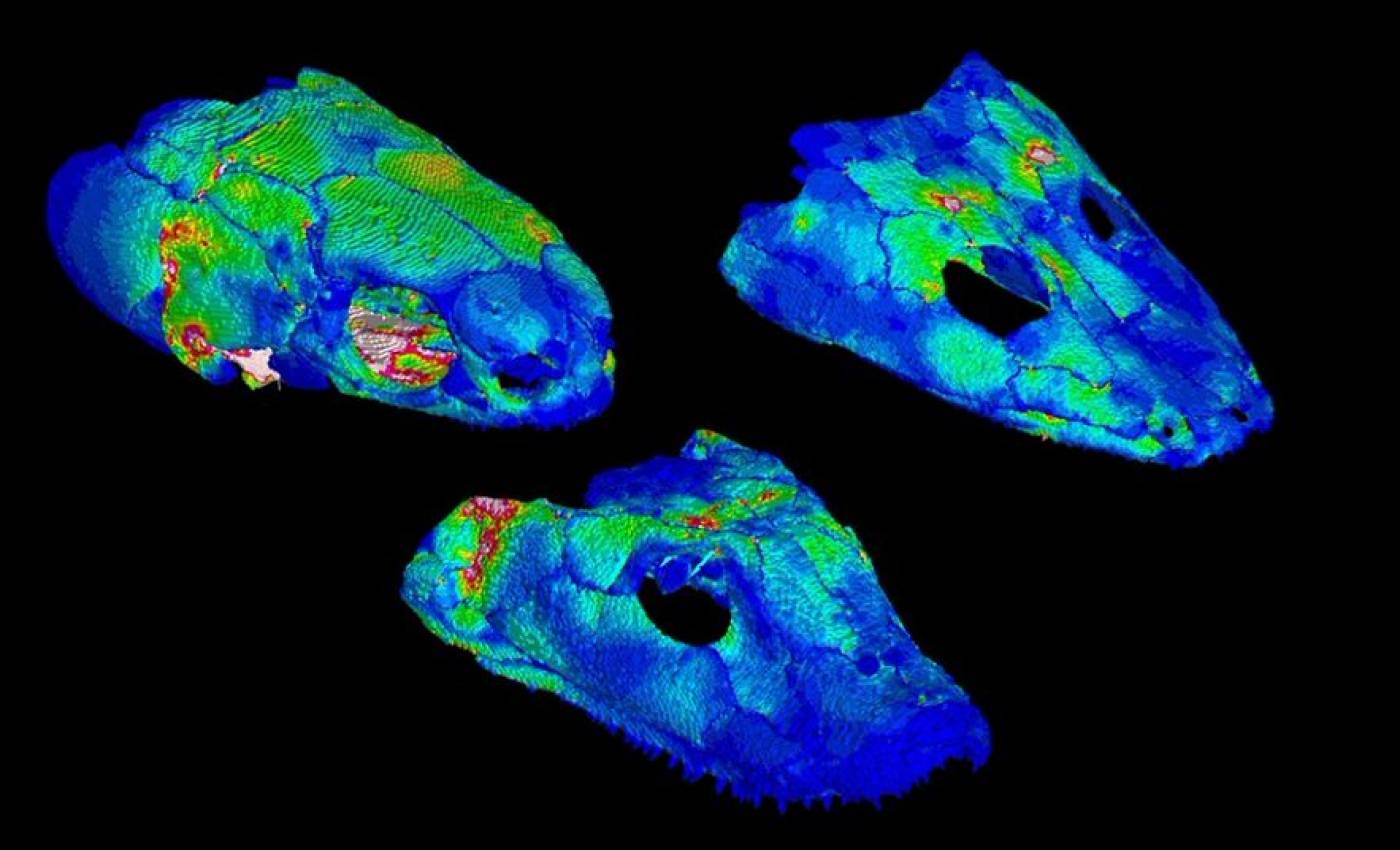
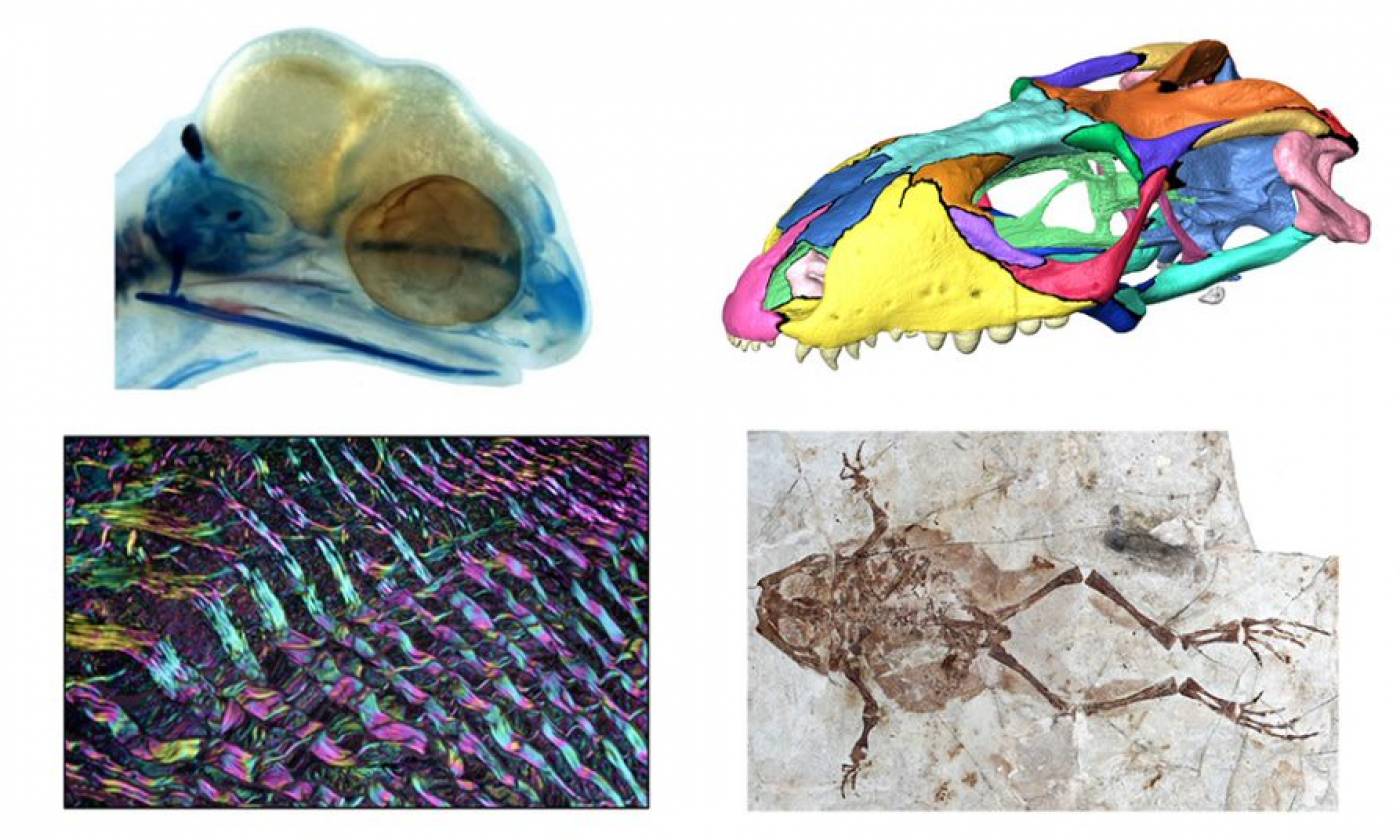
The Porro lab studies the anatomy and biomechanics of living and fossil animals
Work in the Rihel lab is focused on the genes and neurons involved in sleep
The Salinas lab works on synapse formation, plasticity and degeneration in health and disease
The Stern lab explores the processes that establish cell diversity and pattern in the early embryo
The Szabadkai lab studies mitochondrial gene expression and metabolism in health and disease
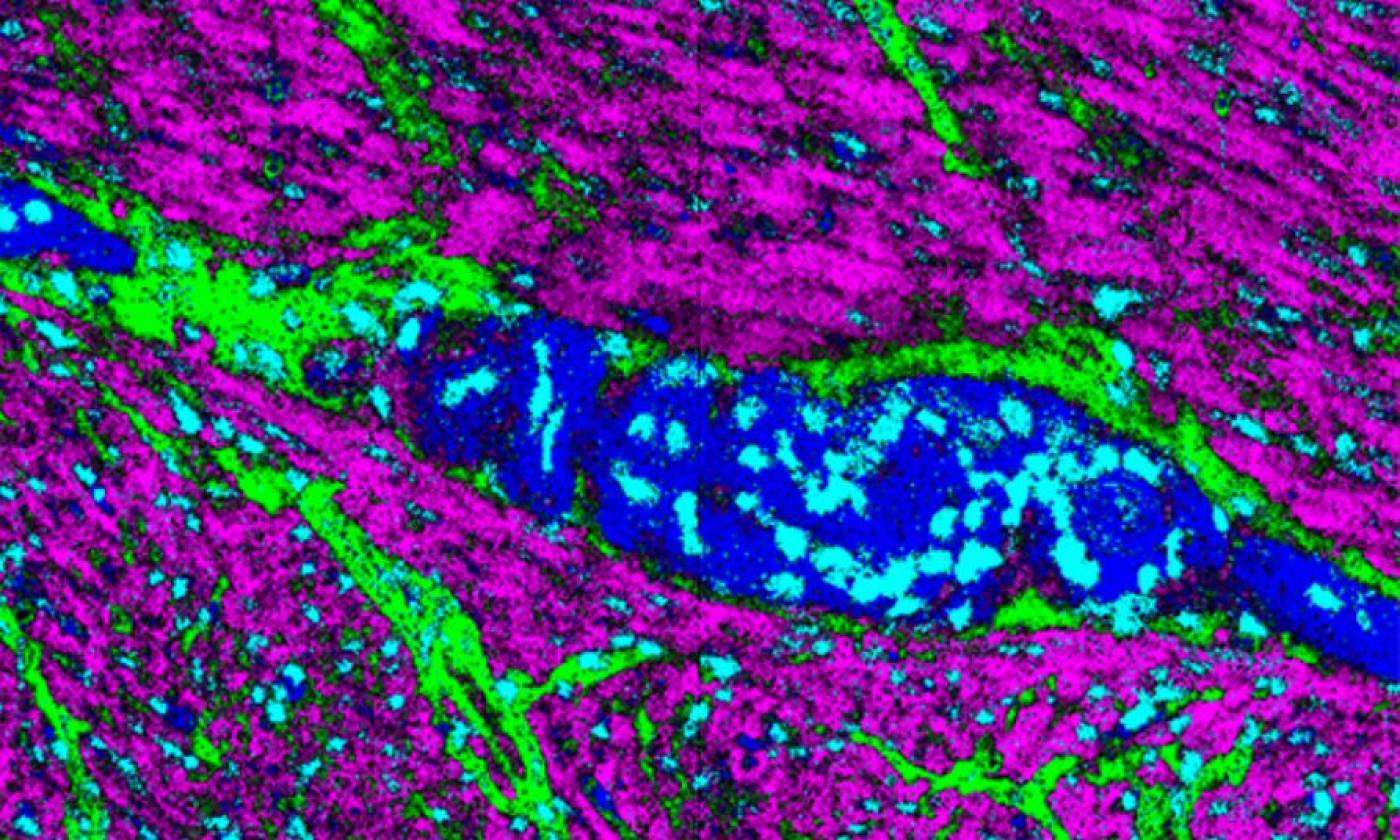
The Thomas lab explores cellular functions across a range of scales from molecules upwards
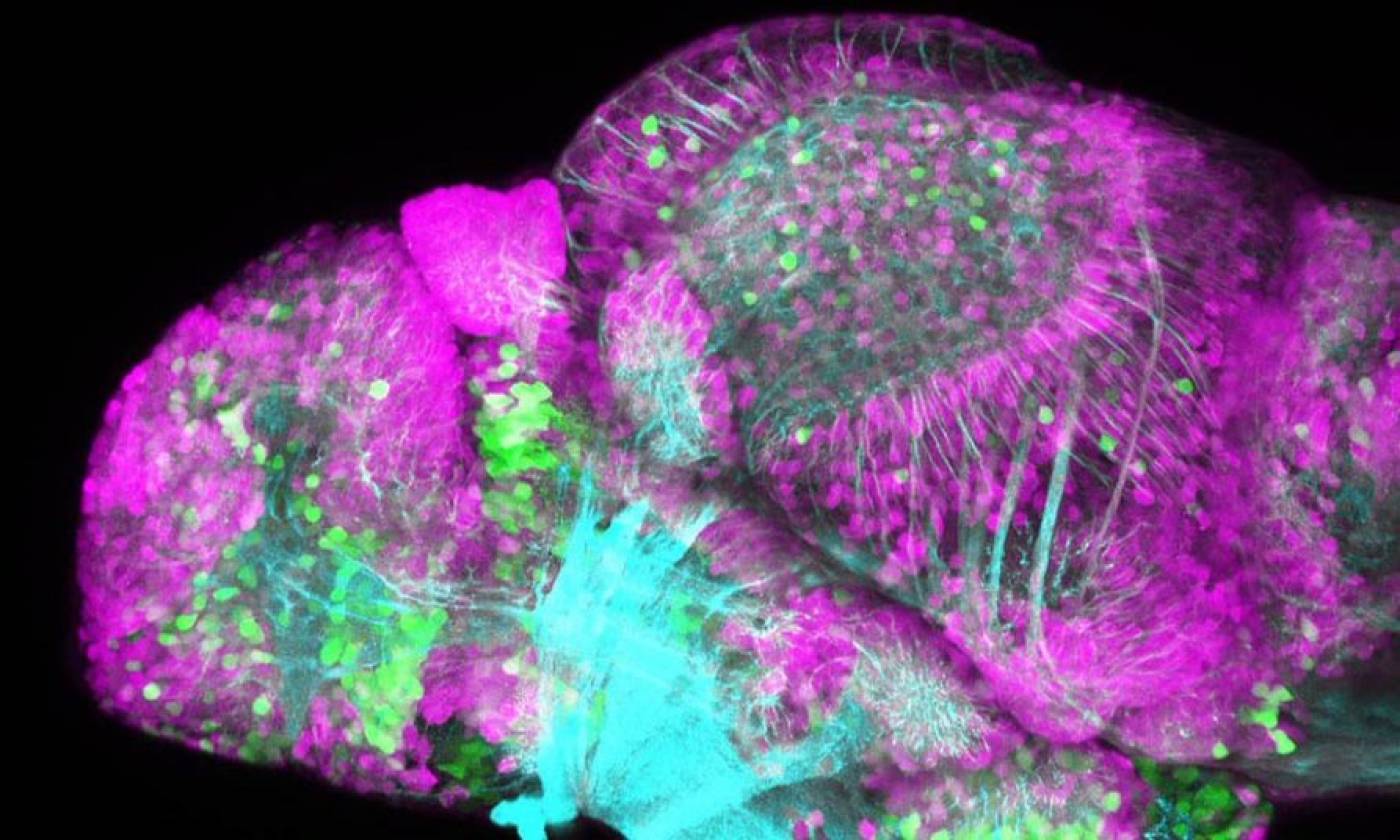
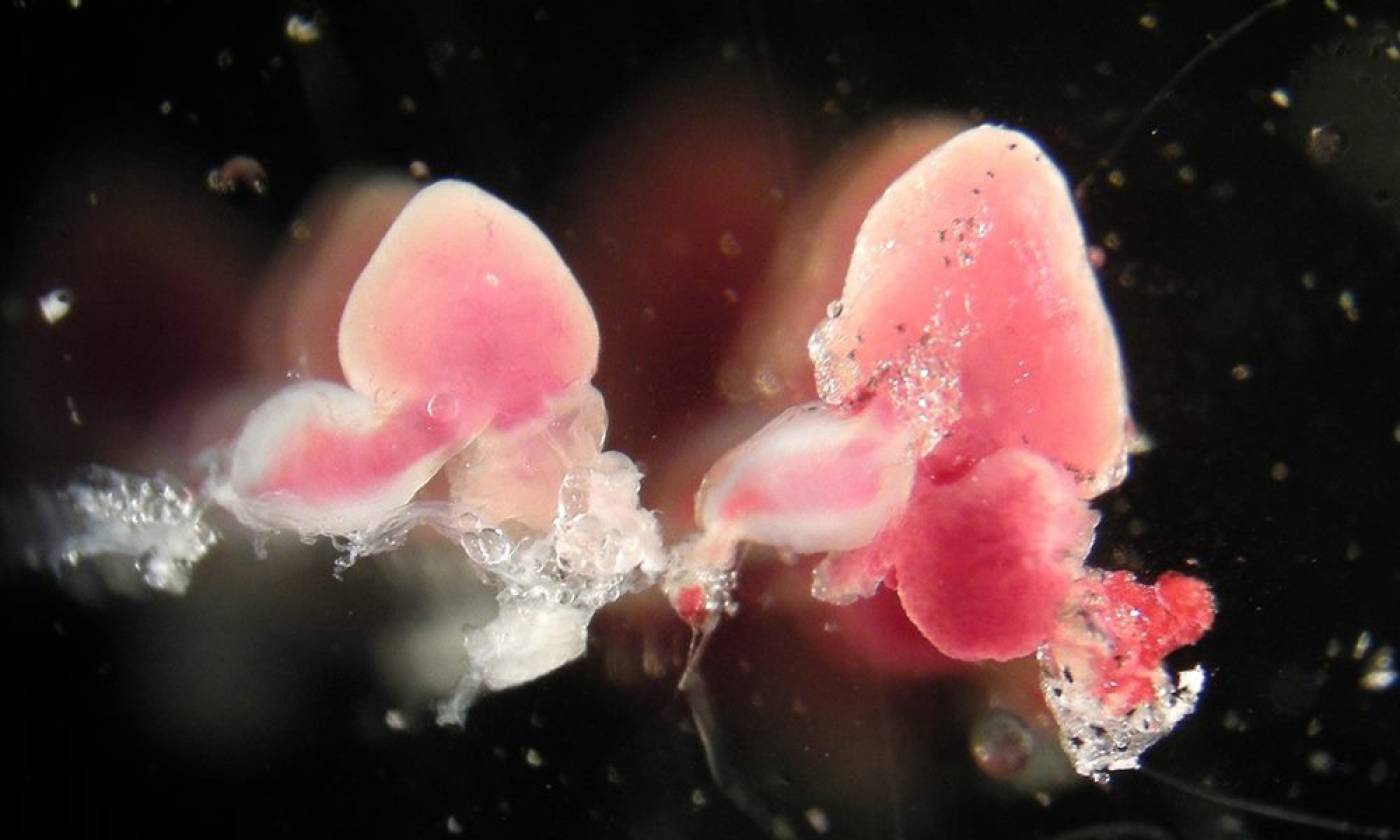
The Yamamoto lab studies evolution and development using cavefish
About
We are a vibrant research department with a long and illustrious history.
Group Leaders
Meet our world-class academic staff and learn about their research.

Study and Training
Information for prospective and enrolled students and postdoctoral researchers.
Centre for Integrative Anatomy (CIA)
The CIA uses diverse and cutting-edge approaches to study anatomy in order to understand structure and function, embryonic development, phylogenetic relationships, ecology and evolution.
Centre for Organelles to Cells to Organisms (COCO)
How do cells truly behave in their natural locale? COCO aims to answer this by integrating studies of cells in vitro with studies in vivo during embryo development.
Consortia and Networks
The Research Department of Cell and Developmental Biology is an active member of the following Consortia and Networks.
Become part of CDB
Establish your lab in a forward thinking collegiate research department.
Postdoc Researchers
Postdocs are a central element of academic life in CDB, find out more here.
Technology Platforms
We host several facilities, equipped with state-of-the-art technology, which support researchers with technical expertise and provide essential services both within the department and across the Division of Biosciences and UCL. These range from our specialist zebrafish facility, through our centres housing cutting-edge microscopes, scanners, computers and related systems to analytical technologies, such as qPCR and Western blot, cloning and D/RNA quantification equipment.