By population structure, population geneticists mean that, instead of a single, simple population, populations are subdivided in some way. The overall "population of populations" is often called a metapopulation, while the individual component populations are often called, well ... subpopulations, but also local populations, or demes. In fact, in many real populations, there may not be any obvious individual populations or substructure at all, and the populations are continuous. However, even in effectively continuous populations, different areas can have different gene frequencies, because the whole metapopulation is not panmictic. For instance, among humans, Scotland, the North of England, and London have some quite major language differences, suggesting substructure, but you would be hard put to find an exact boundary where there is a changeover. Such populations are structured, but continuously, in space.
A very good definition of population structure
is when populations have deviations from Hardy-Weinberg proportions, or
deviations from panmixia. If
there is inbreeding, or selection, or if migration is important, then populations
can be said to be structured in some way.
If populations are subdivided, they can evolve apart, somewhat independently. Population structure allows populations to diversify. This is the reason why population structure is a very important part of evolutionary genetics.
In this lecture we will deal with some simple models of how population structure and migration interferes with natural selection and drift to allow diversification.
Here, several topics are important:
1) Selection
in a patchy environment (this can lead to a kind of
migration/selection balance which favours polymorphism). To avoid interrupting
the flow, we cover this in a separate
page. More is covered under the lecture
Evolution
in space and time).
2) The evolutionary
effect of migration (gene flow) on its own.
3) How to measure
population substructure using
FST.
4) The possibility of drift/migration
balance.
Gene flow (migration)
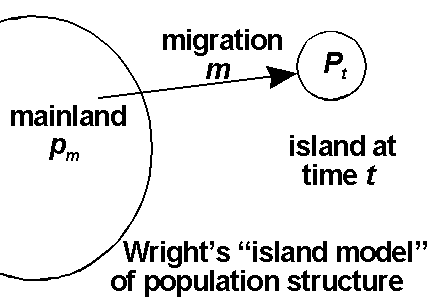

Subdivision into populations with distinct gene frequencies creates a heterozygote deficit due to subdivision into subpopulations (this can be considered a sort of inbreeding):
Example:
AA Aa
aa
HW calcs: pi pi2 2piqi qi2
Deme 1 0.75 0.5625 0.375 0.0625
Deme 2 0.25 0.0625 0.375 0.5625
Totals 1 0.625 0.75 0.625
Overall freqs. 0.5(=pav) 0.3125 0.375 0.3125
pav=fr(AA) + fr(Aa)/2=0.3125 + 0.375/2=0.5; qav=(1-pav)=0.5
To find the heterozygote deficit, F:
2pavqav(1-F)=0.5(1-F)=0.375
Thus:
One can thus measure a sort of inbreeding coefficient, or heterozygote deficit that is due to population subdivision; it is called Fst after Fixation index in the Subpopulation relative to the Total population.F=1-(0.375/0.5)=0.25
Fst
can also be shown to be equal to the standardized variance ![]() of
gene fequency (also called the Wahlund variance) in the
n
subpopulations, divided by (standardized by) the maximum possible variance,
pavqav.
The maximum possible variance is the variance when different populations
are fixed for A or a (pav is the average gene frequency
over all subpopulations). In this case:
of
gene fequency (also called the Wahlund variance) in the
n
subpopulations, divided by (standardized by) the maximum possible variance,
pavqav.
The maximum possible variance is the variance when different populations
are fixed for A or a (pav is the average gene frequency
over all subpopulations). In this case:
Fst=![]() /(pavqav)={[(0.75-0.5)2
+ (0.25-0.5)2]/2} / 0.5x0.5=0.25
/(pavqav)={[(0.75-0.5)2
+ (0.25-0.5)2]/2} / 0.5x0.5=0.25
i.e. the same as we had by looking at the heterozygote deficit. In general it is true that this standardized variance of gene frequencies measures the heterozygote deficit due to subdivision over a number of populations and vice-versa.
Understanding Fst as a fraction of total variance is a useful one. Fst is the proportion of genetic variation found between as opposed to within each populations. Thus (1-Fst) is the proportion of the total metapopulation genetic variation found within as opposed to between populations. If there is a lot of local fixation or inbreeding, Fst will be near 1; if very little, Fst will be low (0.05, for instance, might be a typical value).
Migration (m) from the mainland can balance drift occurring on islands with small population sizes (N);

Fst ~ 1/(1+4Nm). For the proof, CLICK HERE.
PROBLEMS:
Island population structure: very unrealistic!
However, similar equations apply to more
realistic structures, e.g. stepping stone models, continuous populations
with limited migration.
So the island model is much USED (and
also ABUSED!).
EXAMPLES
Population subdivision estimated from allele
frequencies in some wild populations:
Organism No. pops. No. loci Fst Nem High levels of gene flow x population size Tobacco budworm moth (Southeast USA) 60 13 0.002 135 Intermediate levels of gene flow x population size Human (3 major "races") 3 35 0.069 3.4 (Yanomami villages) 37 15 0.077 3.0 (Italian villages) 34 3 0.035 6.9 Low levels of gene flow x population size House mouse 4 40 0.113 2.0 Kangaroo rat 9 18 0.674 0.12
WRIGHT'S "ISOLATION
BY DISTANCE MODEL" OF POPULATION STRUCTURE
(He made a lot of models, didn't he!)
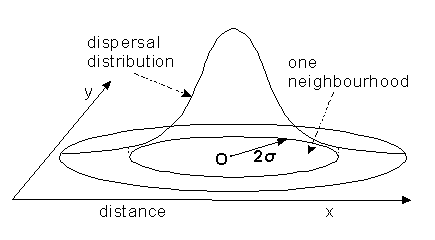
Measuring dispersal
![]() If
the dispersal of an individual between place of birth and breeding site
is essentially random, it resembles a "drunkards walk". You have probably
encountered this sort of movement in physics already; it has the same distribution
as passive diffusion, a two-dimensional normal distribution.
If
the dispersal of an individual between place of birth and breeding site
is essentially random, it resembles a "drunkards walk". You have probably
encountered this sort of movement in physics already; it has the same distribution
as passive diffusion, a two-dimensional normal distribution.
If this is true, dispersal distance can
simply measured as the standard deviation, ![]() ,
of the dispersal distribution. A population "neighbourhood" can be
defined approximately as a group of individuals who come from an area 2
,
of the dispersal distribution. A population "neighbourhood" can be
defined approximately as a group of individuals who come from an area 2![]() wide.
wide.
[Strictly, ![]() is a valid measure of dispersal only if dispersal is exactly normally distributed.
Many field studies have shown that dispersal is actually leptokurtic,
i.e. most offspring breed very close to their parents, but some breed an
enormous distance away. In practice, it doesn't much matter if dispersal
is non-normal, provided it is not too extreme.]
is a valid measure of dispersal only if dispersal is exactly normally distributed.
Many field studies have shown that dispersal is actually leptokurtic,
i.e. most offspring breed very close to their parents, but some breed an
enormous distance away. In practice, it doesn't much matter if dispersal
is non-normal, provided it is not too extreme.]
Neighbourhood population size
The neighbourhood population size was defined
by Wright as the population size in a within a neighourhood of radius 2![]() where population density is d. Therefore, the neighbourhood
population size, Nb = 4
where population density is d. Therefore, the neighbourhood
population size, Nb = 4![]()
![]() 2d.
The neighbourhood population size plays the same role as the value of Nm
in the island model: it determines the amount of variation between populations
at equilibrium between gene flow and drift, measured by FST.
Like Nm, the neighbourhood size is a product of population number
and migration rate. But because the isolation by distance model is
more realistic than the island model (at least, for most situations), it
is potentially more useful. For isolation by distance, we can estimate
population density and dispersal distance, and estimate the value of neighbourhood
size, and hence the expected levels of variation from population to population.
Many people try to apply the island model to spatially extended, continuous
populations, but it wasn't really designed for such use.
2d.
The neighbourhood population size plays the same role as the value of Nm
in the island model: it determines the amount of variation between populations
at equilibrium between gene flow and drift, measured by FST.
Like Nm, the neighbourhood size is a product of population number
and migration rate. But because the isolation by distance model is
more realistic than the island model (at least, for most situations), it
is potentially more useful. For isolation by distance, we can estimate
population density and dispersal distance, and estimate the value of neighbourhood
size, and hence the expected levels of variation from population to population.
Many people try to apply the island model to spatially extended, continuous
populations, but it wasn't really designed for such use.
So today we have covered simple ideas of subdivided populations, and shown how selection in a patchy environment may maintain polymorphisms, even with random mating. We then went on to discuss the evolutionary effects of migration (it homogenizes gene frequencies), discussed how differentiation produces an overall heterozygote deficit (FST) at the metapopulation level, even though there is random mating within subpopulations. If differentiation is caused by genetic drift in small local populations, then this FST measures a kind of inbreeding.
We then discussed how to use this measure to study the equilibrium between drift and migration. The former tends to cause differentiation, the latter to homogenize gene frequencies. This is a drift/migration balance, but you might also imagine situations where the differentiation is produced by patchy selection, producing a selection/migration balance. Geographic selection that is more consistent over larger areas is covered in more depth in Evolution in space and time.
FURTHER READING
FUTUYMA, DJ 1998.
Evolutionary Biology. Chapter 11 (pp. 314-329).