Coot
Coot is one of the best programs written over the last couple years for manual model building, and is constantly having new features added. It is written by Paul Emsley who has also provided a helpful website with both a FAQ and a wiki.
The best way to learn how to use Coot is to experiment yourself, however the following should provide some hints and tips I have found useful.
Coot allows you to manually change your pdb file based upon experimental electron density. If you are using Refmac for refinement coot can directly read your mtz file as Refmac adds phase information to the file. Alternatively you can generate various map files from CNS, SHELX or the ccp4 FFT program and read these in separately.
1) Loading your data
To start coot type:
setccp4 (necessary to load the refinement dictionaries)
setcoot
coot
Coot will then open and often give you a little tip box such as:
You can look through the tips if you like however I normally just close this box.
To load your pdb file use the mouse to select:
File -> Open Coordinates...
and then simply double click on the pdb file which should open in the main graphic window.
If you are using Refmac you can next load the mtz file by selecting:
File -> Open MTZ, mmCIF, fcf or phs
(I think you can select fcf if you have run shelx but I haven't checked this).
You will next get a box that looks like this:
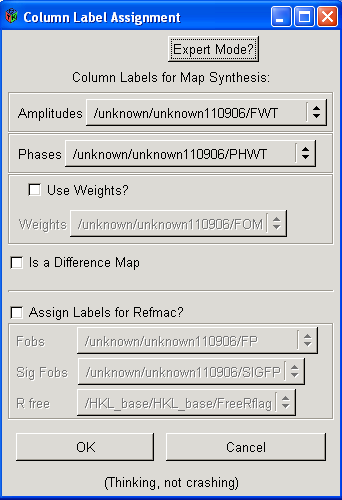
If you want to load a 2Fo-Fc map just click OK. To load your Fo-Fc map simply repeat this procedure BUT select DELFWT and PHDELWT for the Amplitudes and Phases, and then make sure the "Is a Difference Map" box is selected. Just selecting "Is a Difference Map" will not generate an Fo-Fc difference map - make sure you make the appropriate selection for Amplitudes and Phases!
If you are not using Refmac and need to load a separate map file, select:
File -> Open Map...
The map file has to be in ccp4 format and have the extension .map To convert maps from cns or shelx (omap) formats use the usf program "mapman" e.g.:
setccp4
usf mapman
re m1 mymap.map omap
no m1
wr m1 mymap.map ccp4
quit
2) Viewing your model:
Once you have loaded your coordinates, 2Fo-Fc and Fo-Fc maps the screen should look something like this:
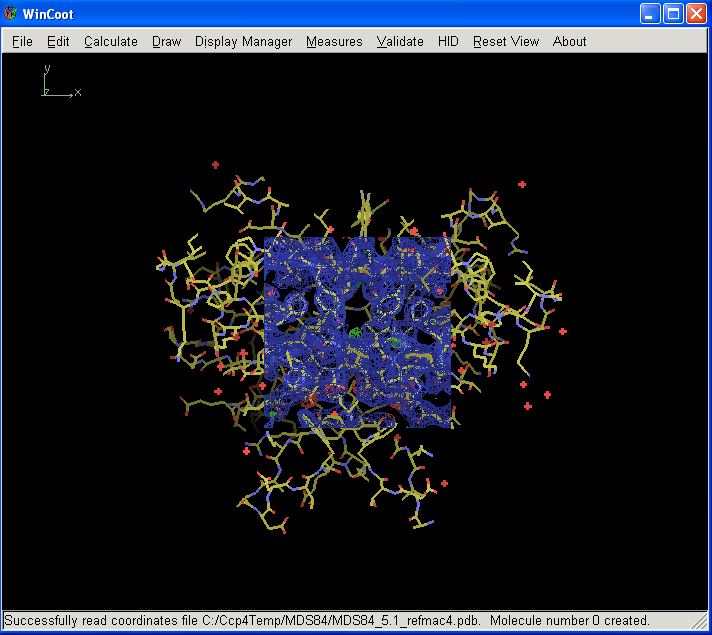
Carbon atoms are coloured yellow, nitrogen's blue, oxygen's and waters red, 2Fo-Fc density dark blue, Fo-Fc positive density green and negative density red.
Coot uses a live mouse which lets you rotate the image by holding down the left mouse button and moving the mouse around the graphics window. You can zoom in and out by using the right mouse button. Centering can be achieved by middle-clicking on an atom. Alternatively you can move the view around by holding down the ctrl key and left mouse button and dragging the mouse around the screen(the center of the view is indicated by a pink box).
To contour the maps select Display Manager from the top menu and then make sure the "Scroll" box is selected for the map you wish to contour. If you then click on the main display the map can be contoured using the wheel on the mouse, or alternatively using the + and - buttons on the keyboard. The contour level is shown in electrons and sigma on the top of the screen. Difference maps contour simultaneously giving your positive and negative electron density at the same contour level (but with reversed sign!).
To select specific residues click on Draw -> Go To Atom This will give you the following box:
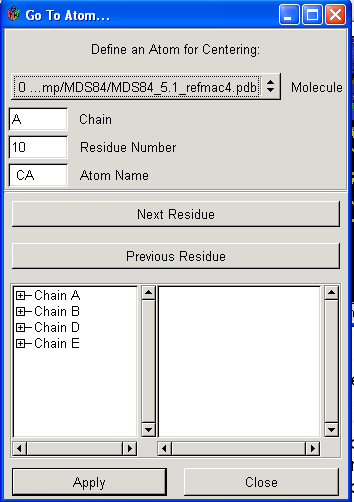
You can either directly specify the atom to jump to using the Chain, Residue Number and Atom name, or alternatively you can click on the "+" next to the different chains and then double click on a residue. Once at a residue you can move to the next residue along a chain by pressing the space bar on your keyboard. Another cool function is the automatic rotate which can be turned on and off using the "I" key on your keyboard.
To turn on symmetry related molecules click Draw -> Cell & Symmetry and then select the box "Master switch: show symmetry atoms?", yes.
3) Editing your model:
In order to make changes to your pdb file you need to bring up the "Model/Fit/Refine" panel by selecting:
Calculate -> Model/Fit/Refine
This will give you a box that should look like this:

As this box can be hidden by the main viewer I normally drag it to the left hand side of my screen and then resize the main view (just by dragging the window from its corners in the normal way) so that I can see all of the viewer and the model/fit/refine screen at the same time. Most of the buttons on Model/Fit/Refine are self-explanatory however I find the most helpful to be:
(NB for both Real Space refine and Regularise zone you need to have the appropriate dictionaries read into memory. This can be achieved by typing "setccp4" BEFORE you open coot. If you have a ligand you will need to read the "Refmac" dictionary for that ligand into coot. Create the dictionary using PRODRG2 and then select File -> Import CIF dictionary. Make sure your dictionary was made using the same ligand residue ID as in your pdb file.)
By altering the sliders you can move your selection around the screen. This function often works very well if you need to move something like an entire ligand. Once you have positioned the segment it is best to use the "Regularise zone" function to correct the geometry.
The rest of the boxes in the Model/Fit/Refine window are generally self-explanatory. Try clicking on them to see what they do! The final button "Run Refmac" allows you to run Refmac on your edited model, however I prefer to save my coordinates (File -> Save Coordinates...) and then run Refmac from the ccp4i GUI to have more control over the parameters.
4) Validation Functions:
There are a certain number of validation functions included in the "Validate" menu. The more useful functions include a Ramachandran plot that allows you to click on residues outside the allowed regions, and both a geometry and temperature factor analysis that shows you to go problematic residues by clicking on the amber or red bars. Note, however, that both temperature and geometry analysis has to be "re-set" by reloading your pdb file (eg after a refinement round) so just because the red/amber bar has disappeared once you have edited the residue does not mean you have fixed the problem. Also expect the terminal residues to be flagged as "problems", however these can generally be ignored.
Do check out the other functions on this menu, although it might be worth reading up about what they are looking at.
5) Other Useful functions:
Coot is a huge program with a long instruction manual and lots of online tutorials/talks. This webpage gives only the extreme basics because it is probably much better to learn for yourself using trial and error. However, the following represents a list (which I will endeavour to expand) of various useful functions I have come across: