 Close
Close

John Collinge
 Tel: 020 7837 4888 Courtauld Building, Room 504 UCL Profile • PubMed • |
|
Research Synopsis
Prions are of major interest in biology. Not only are they infectious agents capable of entering and colonising a host and evading its defences while being composed essentially of a single polypeptide, but the existence of multiple prion strains that can maintain their identity when serially passaged in a range of hosts constitutes protein-based inheritance: both aspects raising fascinating evolutionary questions. It is becoming clear that prion strains constitute an ensemble or quasispecies, with interplay between propagating and toxic entities, phenomena which may cast light on other diseases associated with seeded protein polymerisation. The last decade has seen an explosion of international interest in the possible wider relevance of these molecular processes in human disease. The rapidly developing field of ‘protein-misfolding diseases’ has prion disease as a key paradigm. The commonest neurodegenerative diseases can be considered in this category, notably Alzheimer’s disease. While ‘prion-like’ seeding of protein aggregates from several neurodegenerative diseases has been experimentally reproduced in animal models, and we have recently published evidence that seeding of Aβ pathology may have occurred in humans, its full pathogenetic role and significance to the natural diseases remains to be elucidated. We are utilising our expertise to develop analogous experimental tools to those successfully employed in prion disease to investigate these phenomena in other diseases.
We see prion diseases as providing a ‘way in’ to tackle degenerative dementia, many years of work having led to a sufficient understanding of fundamental mechanisms, and development of the necessary molecular and cellular tools and animal models, to enable the development of rational therapeutics. The rapidly progressive nature of CJD, allied with our intensive longitudinal clinical studies and development of unique biomarkers, enable us to test drugs rapidly and efficiently. Essentially, we see development of treatments of CJD, although a massive goal in itself which will transform the lives of people affected by these distressing diseases, as likely to be highly informative in wider neurodegeneration therapeutics research. No disease progression-modifying treatment exists yet for any neurodegenerative dementia and an effective treatment for prion diseases, despite its relative rarity, would be transformational in dementia research much more widely.
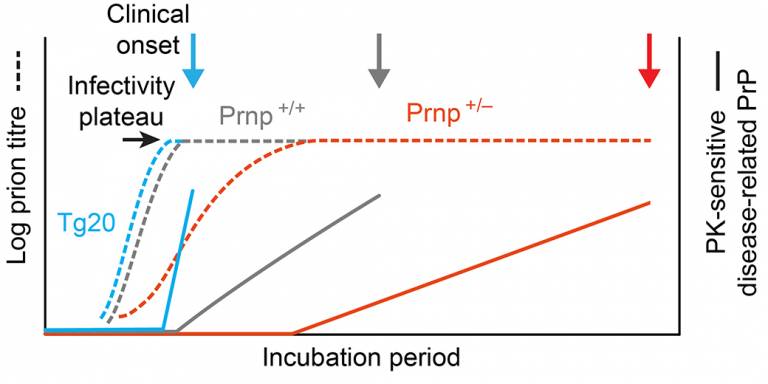
Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Sandberg MK, Al-Doujaily H, Sharps B, Clarke AR, Collinge J Nature. 2011; 470: 540-2. doi:10.1038/nature09768
Prion neuropathology follows the accumulation of alternate prion protein isoforms after infective titre has peaked. Sandberg MK, Al-Doujaily H, Sharps B, De Oliveira MW, Schmidt C, Richard-Londt A, Lyall S, Linehan JM, Brandner S, Wadsworth JD, Clarke AR, Collinge J. Nat Commun. 2014; 5: 4347. doi:10.1038/ncomms5347
Peripheral administration of a humanized anti-PrP antibody blocks Alzheimer's disease Abeta synaptotoxicity.Klyubin I, Nicoll AJ, Khalili-Shirazi A, Farmer M, Canning S, Mably A, Linehan J, Brown A, Wakeling M, Brandner S, Walsh DM, Rowan MJ, Collinge J. J Neurosci. 2014; 34: 6140-5. doi:10.1523/JNEUROSCI.3526-13.2014
Mammalian prions and their wider relevance in neurodegenerative diseases. Collinge J. Nature. 2016; 539: 217-26. doi:10.1038/nature20415
Transmission of amyloid-beta protein pathology from cadaveric pituitary growth hormone. Purro SA, Farrow MA, Linehan J, Nazari T, Thomas DX, Chen Z, Mengel D, Saito T, Saido T, Rudge P, Brandner S, Walsh DM, Collinge J. Nature 2018; 564: 415-19. doi:10.1038/s41586-018-0790-y
Highly infectious prions are not directly neurotoxic Benilova I, Reilly M, Terry C, Wenborn A, Schmidt C, Marinho AT, Risse E, Al-Doujailya H, Wiggins De Oliveira M, Sandberg MK, Wadsworth JDF, Jat PS, Collinge J. PNAS September 22, 2020 117 (38) 23815-23822; first published September 8, 2020; https://doi.org/10.1073/pnas.2007406117