 Close
Close

Discovery of new ALS and dementia disease mechanism raises treatment hopes
23 February 2022
A pioneering new study led by UCL and National Institutes of Health (NIH) scientists has revealed, for the first time, why a common genetic variant worsens disease outcomes for people with the devastating adult-onset neurodegenerative diseases ALS and FTD.
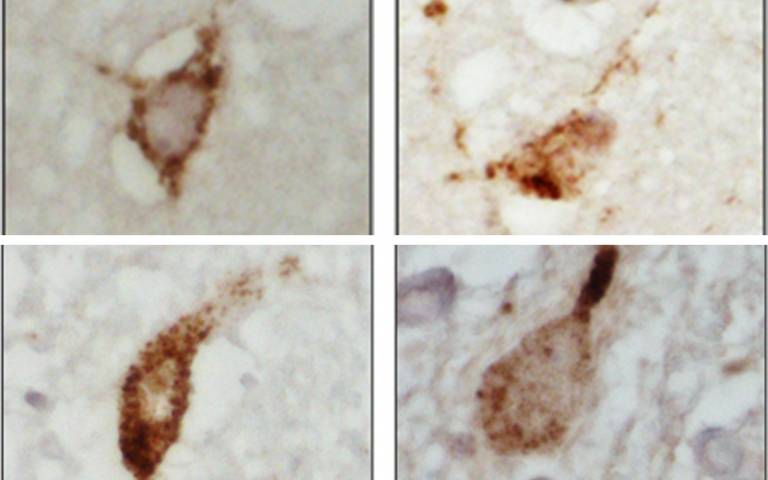
Published in Nature, the study shows how TDP-43 protein depletion, associated with almost all cases (97%) of vALS amyotrophic lateral sclerosis (ALS) and half of frontotemporal dementia (FTD) cases, corrupts the genetic instructions for the critical neuronal protein UNC13A.
Strikingly, it found that a mysterious genetic variant previously associated with disease risk increases the chance of UNC13A’s genetic instructions being corrupted among people with the diseases, thereby worsening risk and severity of ALS and FTD.
UNC13A enables neurons (nerve cells) to communicate with each other via neurotransmitter release, and data from animal models suggests its loss from neurons can be fatal. The researchers believe that the corruption of UNC13A’s genetic instructions in patients may have similarly harmful consequences.
ALS is the most common motor neuron disease and there is no known cure; it affects the brain and spinal cord by attacking the neurons and nerves which control movement, causing them to die. There is currently only one approved drug for ALS in the UK, which extends lifespan by a few months, and is only effective for a tiny minority of patients. One third of patients die within one year of diagnosis.
FTD is a related disease with similar underlying causes; symptoms include language impairment, changes in personality and cognitive difficulties.
Researchers say the discovery raises hope for new treatments; by developing a therapy that blocks the corruption of UNC13A’s genetic instructions, disease progression could be slowed for most people with ALS and around half of patients with FTD.
Corresponding author Professor Pietro Fratta (UCL Queen Square Motor Neuron Disease Centre, UCL Queen Square Institute of Neurology) said: “The majority of research into gene therapy has focused on genes implicated in familial ALS (patients with a family history of the disease), but the vast majority of ALS cases are sporadic, with no known family history.”
Co-corresponding author Dr Michael Ward (National Institute of Neurological Disorders and Stroke, NIH, US) added: “We have known for a long time that genetic variants in UNC13A cause an increased risk of ALS and dementia, but nobody had figured out why this is the case. Together, our teams showed exactly how this genetic risk factor for ALS interplays with the core disease mechanism, TDP-43 loss, in order to worsen the disease course.”
TDP-43 – a key player in ALS and FTD
Arguably the most important protein in ALS research is TDP-43, as in most cases (as well as half of FTD cases), the protein is incorrectly ejected from the cell’s nucleus. This prevents TDP-43 from performing its important functions, such as ensuring that mRNA is produced correctly.
Dr Ward said: “We have known for a long time that most patients with ALS, and about half of FTD patients, lose the function of a key protein called TDP-43, wreaking havoc in nerve cells that are affected. But we haven’t known how to reverse the most problematic consequences of TDP-43 loss.”
As part of the experimental study, the researchers used skin-derived human stem cells to make neuronal cells in dishes and removed the TDP-43 protein from these cells using a new technology based on CRISPR-Cas9, the Nobel-prize winning gene-editing technology.
The scientists were then able to study how these neurons without TDP-43 differed from healthy neurons. They found that the mRNAs for the UNC13A protein were corrupted, meaning the ribosomes in the lab-grown neurons were unable to correctly produce the UNC13A protein. Furthermore, when the team looked at ALS and FTD patient brain samples, they again found that the mRNAs for UNC13A were incorrect, confirming that their experiments replicated the real-world disease process.
Given the essential role UNC13A plays in facilitating neuron communication, its corruption is thus likely to impair neuronal function and contribute to neurodegeneration in those with ALS and FTD.
Genetic variants increase the risk of UNC13A mRNA corruption
The UNC13A gene and its corresponding protein are of longstanding interest to motor neuron disease and FTD researchers, with previous studies showing common genetic variants increase the risk and severity of the diseases, despite being benign in most people unaffected by the diseases (half the population carries one of these variants, which are only harmful in people with ALS or FTD). However, despite over a decade of research, the exact reason for this has remained mysterious, as these variants do not directly alter the UNC13A protein-coding sequence, but are instead located in a region of ‘junk DNA’.
The researchers believe they have uncovered the answer to this critical question: they found that the risk-linked variants greatly increase the chance of the UNC13A mRNA becoming corrupted once the ALS and FTD disease course, and the associated loss of TDP-43 protein, has begun. Thus, patients with these genetic variants are likely to suffer greater loss of UNC13A, resulting in more severe disease.
Co-lead author, PhD student Oscar Wilkins (UCL Queen Square Institute of Neurology and Francis Crick Institute), said: “These results represent a significant breakthrough for several reasons. Firstly, they explain why UNC13A genetic variants increase the risk of motor neuron disease and dementia, a question which has puzzled researchers for over a decade. They are also the first to demonstrate a genetic link specifically between loss of nuclear TDP-43 function and ALS, improving scientific understanding of this central disease mechanism.”
Next steps
Professor Fratta said: “We have built on years of genetic research that identified that UNC13A was implicated in motor neuron disease and FTD, and supported it with a new molecular biology finding that confirms that the gene is absolutely fundamental to the disease process.
“We are hoping to carry out trials over the coming years to develop such a treatment that could potentially greatly improve the lives of people living with ALS.”
The researchers are confident that with this new information, new therapies for motor neuron disease can be created that stop UNC13A mRNAs from being corrupted in patients.
The study involved researchers at UCL, NIH, the Crick, New York Genome Center, Icahn School of Medicine at Mount Sinai, International Centre for Genetic Engineering and Biotechnology, and the National Institute of Chemistry (Slovenia). The study was funded by the Medical Research Council and the Motor Neurone Disease Association and the NIH with support from Rosetrees Trust, The Robert Packard Center for ALS Research, Wellcome, Collaborative Centre for Applied Nanotechnology and Collaborative Center for X-linked Dystonia-Parkinsonism.
Links
- Research paper in Nature
- Professor Pietro Fratta’s academic profile
- Fratta lab
- UCL Queen Square Motor Neuron Disease Centre
- UCL Queen Square Institute of Neurology
Image
Abnormal aggregations of the TDP-43 protein, seen in neurons affected with ALS. Credit: Alex Bampton and Professor Tammaryn Lashley, UCL Queen Square Institute of Neurology, co-authors of the Nature paper.
Media contact
Chris Lane
Tel: +44 (0)20 7679 9222
Email: chris.lane [at] ucl.ac.uk
Follow us