|
|
Scott Woodley is a Professor of Computational Chemistry and Physics within the Department of Chemistry at UCL and Deputy Director of the Molecular Modelling and Materials Science (M3S) Centre for Doctoral Training (CDT).
His research is focused on the structures and properties of materials.
Scott develops, optimises and employs state-of-the-art materials software that is based on atomic and electronic levels of theory (either semi-empirical or ab initio).
|
|
|
|
Enabling Activities
A significant proportion of his time is devoted to helping the materials modelling community by helping UK academics gain access to computer resources and networking, including:
1. Two terms on the CCP5 executive committee (2007-10 and 2016-19) during which he organised the CCP5 annual conferences at the LSE in 2008 (with Profs. Fernando Bresme and Dorothy Duffy) and in 2009 (when the focus was switched from MD to MC and static lattice techniques) and is currently organising this year's 2019 annual meeting (with Drs Alin Marin Elena and Kostya Trachenko); and helped to secure funding for CCP5 (post 2010) and for the development of DL_POLY_4 (rare events, with Prof. Mark Rodgers).
2. One term on the BACG executive committee (2010-16) during which he organised the BACG annual conferences at UCL in 2010 (with Prof. Nora de Leeuw) and at Queen Mary in 2016 (with Dr Devis Di Tommaso).
3. Part of EPSRC procurement team for UK's HEC national resource (ARCHER), and more recently of the Project Working Group and, currently, chair of the benchmarking team for ARCHER's replacement.
4. Manager and current chair of UK's largest HEC consortium, the Materials Chemistry Consortium (MCC), during which he has secured EPSRC funding (as Researcher Co-I, Co-I and, more recently, PI) for this community. Present funding includes compute resources on both ARCHER and THOMAS, as well as vital COSEC support for development of CRYSTAL, DL_POLY, DL_MONTE and CHEMSHELL. Scott organises two full one-day meetings per year for the MCC allocation of national computer resources, and the MCC 3-day conferences which were held in London 2012, Cardiff 2016 (with co-organiser, Dr David Willock) and Lincoln 2018 (with co-organiser Drs Arun Chutia and Matthew Watkins).
5. Co-I for HEC THOMAS, member of management and users committees, and point of Contact for UCL users.
6. Developer and manager of the web-database of Published Interatomic Potential Parameters.
Active Research Activities
His group is multidisciplinary and currently consists of 2 research associates, 3 PhD students and 3 MRes students. Projects include:
1. Development of new interatomic potential models for sp-lone pair bearing cations. It is planned that this model will be made available to the modelling community via its implementation within the popular General Utilities Lattice Package (GULP), which Scott has helped develop new functionality for since 1997.
2. Development of global optimisation routines within the KLMC software and its application to predicting structures of nanoclusters, nanotubes and nanowires.
3. The SAINT project: we are developing a software suite that will support advanced and complex surface and interface simulations of materials employed across the fields of energy, catalysis and life sciences. Surfaces and interfaces of materials control important processes, including corrosion, catalysis, electronic and photovoltaic device operation. Surface and interface studies pose greater challenges than the respective bulk investigations, but a wealth of new experimental techniques to interrogate surfaces need now to be fully complemented with tools for their systematic investigation in silico. Work packages include development of (a) new searchable web-database for surface models; (b) web-tools for creating surface models and running CRYSTAL simulations; (c) correction software of unwanted images of defects; and new functionality within (d) CHEMSHELL and (e) CP2K.
Published Research Activities
Scott has published his research in approximately 100 research articles, including several book chapters and co-edited "Frontiers of Nanoscience, Volume 12, Computational Modelling of Nanoparticles, Elsevier" with Prof. Stefan Bromley. Research projects already published include:
|
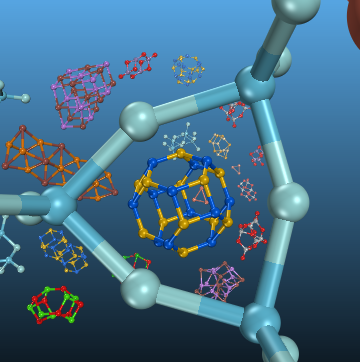
1. To address the question "What is the best or most relevant global minimum for nanoclusters?", we have created, and have reported details of, an open-access database of cluster structures with a web-assisted interface and toolkit as part of the WASP@N project. The HIVE database establishes a map of connectivities within each structure, the information about which is coded and kept as individual labels, called hashkeys, for the nanoclusters. These hashkeys are the basis for structure comparison within the database, and for establishing a map of connectivities between similar structures (topologies). The database is successfully used as a key element in a data-mining study of (MX)12 clusters of three binary compounds (LiI, SrO and GaAs) of which the database has no prior knowledge. The structures are assessed on the energy landscapes determined by the corresponding bulk interatomic potentials. Global optimisation, using a Lamarckian genetic algorithm, is used to search for low lying minima on the same energy landscape to confirm that the data-mined structures form a representative sample of the landscapes, with only very few structures missing from the close energy neighbourhood of the respective global minima.
|

Publication (Faraday Discussions) DOI:10.1039/c8fd00060c
|
|
Publication (Nanoscale) DOI:10.1039/c6nr09072a
|
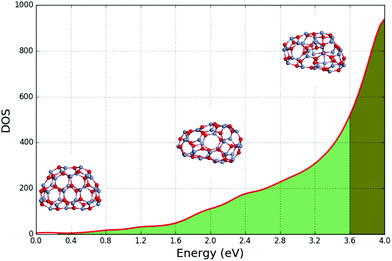
2. We have developed and implemented a new global optimization technique based on a Lamarckian genetic algorithm with the focus on structure diversity. The key process in the efficient search on a given complex energy landscape proves to be the removal of duplicates that is achieved using a topological analysis of candidate structures. The careful geometrical prescreening of newly formed structures and the introduction of new mutation move classes improve the rate of success further. The power of the developed technique, implemented in the Knowledge Led Master Code, or KLMC, is demonstrated by its ability to locate and explore a challenging double funnel landscape of a Lennard-Jones 38 atom system (LJ38). We apply the redeveloped KLMC to investigate three chemically different systems: ionic semiconductor (ZnO)1�32, metallic Ni13 and covalently bonded C60. All four systems have been systematically explored on the energy landscape defined using interatomic potentials. The new developments allowed us to successfully locate the double funnels of LJ38, find new local and global minima for ZnO clusters, extensively explore the Ni13 and C60 (the buckminsterfullerene, or buckyball) potential energy surfaces.
|
|
|
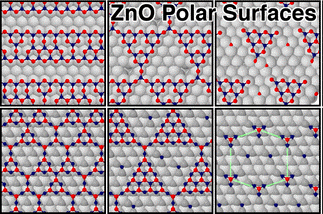
3. We have probed and rationalized the complex surface chemistry of wurtzite ZnO by employing interatomic potential calculations coupled with a Monte Carlo procedure that sampled over 0.5 million local minima. From the predicted structure and stability of the (0001) and (000-1) ZnO surfaces, we have rationalized previous patterns found in STM images and explaining the (1×1) periodicity reported by LEED analysis. The full range of Zn/O surface occupancies was covered for a (5×5) supercell, keeping |mZn - mO|/N ~ 0.24 where m and N are the numbers of occupied surface sites and total surface sites, respectively. Our calculations explain why the (5×5) reconstructions seen in some experiments and highlight the importance of completely canceling the inherent dipole of the unreconstructed polar surfaces. The experimentally observed rich reconstruction patterns can be traced from the lowest occupancy, showing the thermodynamically most stable configurations of both polar surfaces. Triangular and striped reconstructions are seen, inter alia, on both polar surfaces, and hexagonal patterns also appear on the O terminated surface. Our results explain the main experimental structures observed on these complex surfaces. Moreover, grand canonical simulations of ZnO polar surfaces reveal that disorder is favored and, thus, configurational entropic factors is the the cause of their stability.
|
ACS Publication (Chem.Mater) DOI:10.1021/acs.chemmater.7b01487
|
|
Publication (Adv.Mater) DOI:10.1002/adma.201401858
|
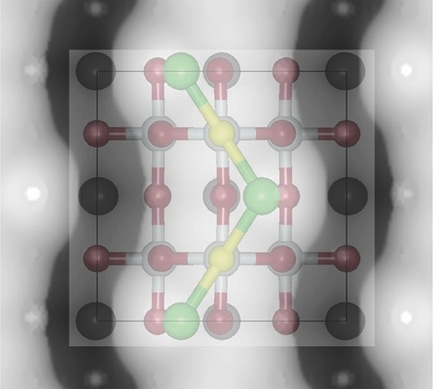
4. Global optimization is used to study the structure of the polar KTaO3 (001) surface. It is found that cation exchange near the surface leads to the most stable structure. This mechanism is likely to be general to metal oxides containing cations of differing charge. This study employed the KLMC software to automate the global search for plausible low energy configurations by: (a) generating the initial models; (b) calling GULP to perform the local optimisations, or structural relaxations; (c) extract and process the required data from GULP output files. All plausible models were refined using VASP.
|
|
|
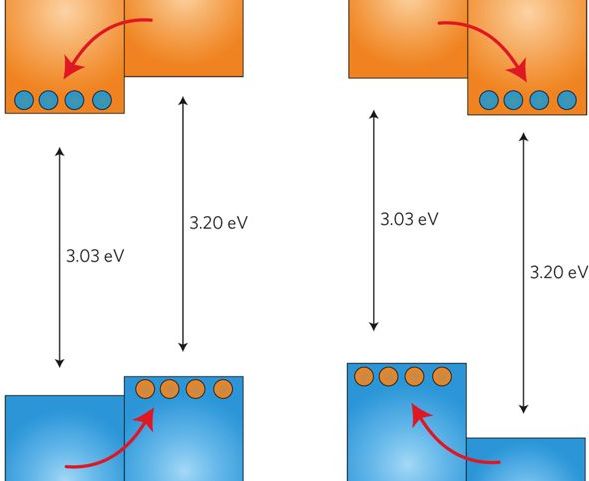
5. The most widely used oxide for photocatalytic applications owing to its low cost and high activity is TiO2. The discovery of the photolysis of water on the surface of TiO2 in 1972 launched four decades of intensive research into the underlying chemical and physical processes involved. Despite much collected evidence, a thoroughly convincing explanation of why mixed-phase samples of anatase and rutile outperform the individual polymorphs has remained elusive6. One long-standing controversy is the energetic alignment of the band edges of the rutile and anatase polymorphs of TiO2. We demonstrated, through a combination of state-of-the-art materials simulation techniques and X-ray photoemission experiments, that a type-II, staggered, band alignment of ~ 0.4eV exists between anatase and rutile with anatase possessing the higher electron affinity, or work function. Our results help to explain the robust separation of photoexcited charge carriers between the two phases and highlight a route to improved photocatalysts.
|
Publication (Nat.Mater) DOI:10.1038/NMAT3697
|
|



 +44 (0)20 7679 4650
Copyright © 1999-2009 UCL
Disclaimer |
Accessibility |
Privacy |
Advanced Search |
Help
+44 (0)20 7679 4650
Copyright © 1999-2009 UCL
Disclaimer |
Accessibility |
Privacy |
Advanced Search |
Help