 Close
Close

New study solves decades old enigma in mitochondrial DNA disorders & advances prospect of treatments
6 December 2021
An international team led by researchers at UCL Queen Square Institute of Neurology report in Nature Communications the identification of small molecules that purge cells of mutant mitochondrial DNA molecules thereby restoring mitochondrial function.
Mitochondria are known as the powerhouses of the cell, and their small circles of DNA are essential to produce energy from the food we eat. Consequently, mutant mitochondrial DNAs are responsible for a plethora of devastating and incurable human diseases. An international team led by researchers at UCL Queen Square Institute of Neurology report in Nature Communications the identification of small molecules that purge cells of mutant mitochondrial DNA molecules thereby restoring mitochondrial function. Exceptionally, this is akin to a 'gene therapy' with small molecules. The findings are of considerable importance as they represent a possible treatment for the future.
Over 30 years ago, researchers at the Queen Square Institute of Neurology (QS IoN) identified the first human diseases caused by mutations in the DNA inside the mitochondria (1). Since then, hundreds of mutations in the mitochondrial DNA (mtDNA) have been associated with a plethora of diseases affecting almost every organ of the body, at any stage of life. Current treatments are only symptomatic and there are no disease-modifying therapies for mtDNA diseases. Antonella Spinazzola of QS IoN and the surviving member of the original team, Ian Holt, together with collaborators at the Wellcome Centre for Mitochondrial Research, Newcastle University and an enthusiastic and dedicated team of young scientists, have discovered a means to counteract mitochondria that harbour mutant mtDNAs.
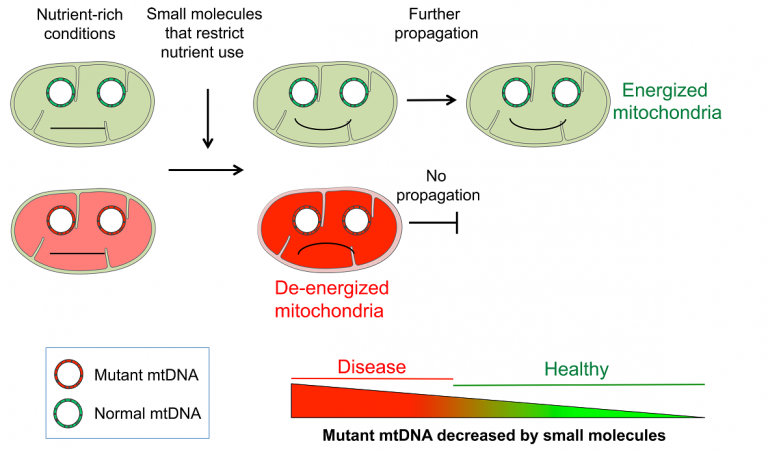
A typical cell contains hundreds of mitochondria each with several copies of mtDNA. Many patients with mitochondrial DNA disorders carry a mixture of mutant and normal mtDNAs, and it has long been a puzzle why the mutant mitochondria persist and even thrive, which is the opposite of Darwinian ‘selection of the fittest’. The new study shows that the defective mitochondria ‘steal’ energy and other resources from the rest of the cell; as such, they behave like parasites. However, when resources (nutrients) are in short supply, and each and every mitochondrion has to ‘fight for itself’, the defective mitochondria are disadvantaged. This insight enabled the researchers to demonstrate that healthy mitochondria can be selected with chemicals that restrict nutrient metabolism and dramatically inhibit the replication of the mutant mtDNAs (2).
The small molecules targeting nutrient metabolism are potential new drugs that are equivalent to a gene therapy, in that they act specifically on the mutant mDNAs, albeit without directly interacting with them. The team, which includes Professor Hanna and Dr. Pitceathly at UCL Queen Square Institute of Neurology, and Professors Taylor and McFarland from Newcastle University, is already building on the new findings, setting up an experimental medicine study as the next step towards developing the small molecules as treatments for these currently incurable diseases.
Professor Antonella Spinazzola, senior author of the study, stated: “Discovering that nutrient levels can have such a dramatic effect on the replication of mutant mtDNA, and that we can manipulate them with the small molecules, represent two major steps towards understanding and developing treatments for a group of human diseases we have been wrestling with for decades".
Co-senior author, Professor Ian Holt, said: “As well as achieving our long-term goal of identifying potential drugs that specifically inhibit the replication of mutant mitochondrial DNAs, we have opened up a new area of research: the regulation of mitochondrial DNA metabolism, and thus mitochondrial energy production and cell function, via the manipulation of nutrients”.
Professors Robert Taylor and Robert McFarland from Newcastle University added: “In identifying the cellular metabolic constraints that can influence mitochondrial DNA replication, this ground-breaking study not only resolves a long-standing conundrum in the molecular pathology of mitochondrial disease, but offers an exciting opportunity for rapid translation of this discovery into real patient benefit through repurposing a compound that has already been tested in human subjects.”
Professor Michael Hanna, Director of UCL Queen Square Institute of Neurology commented:“ Since the original discovery that mtDNA deletions are linked to human disease at the Queen Square Institute of Neurology, we have been striving for a deeper understanding of diseases mechanisms and ways to develop therapies for these devastating disorders affecting children and adults. This breakthrough paves the way for an exciting new chapter of clinical experimental research using agents which target and reduce the number of dysfunctional mitochondria”.
All the authors are extremely grateful to all the funders, patients and families that over the years have supported the research leading to this achievement.
Links
- Holt, I., Harding, A. & Morgan-Hughes, J. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 331, 717–719 (1988)
- Pantic, B., Ives, D., Mennuni, M. et al. 2-Deoxy-D-glucose couples mitochondrial DNA replication with mitochondrial fitness and promotes the selection of wild-type over mutant mitochondrial DNA. Nat Commun 12, 6997 (2021). https://doi.org/10.1038/s41467-021-26829-0
- Professor Spinazzola‘s academic profile
- Professor Holt's Ikerbasque profile
- Professor Holt's Biodonostia profile
Image
Although mitochondria with mutant mitochondrial DNA (mtDNA) do not function properly, in nutrient-rich conditions they can survive by taking energy and resources from the rest of the cell. However, when nutrient utilization is restricted with chemicals, such as 2-Deoxy-D-glucose, defective mitochondria are unable to support themselves, and only the mitochondria with normal mtDNA can prosper.