 Close
Close

Non-ketotic hyperglycinemia (NKH), or glycine encephalopathy, is a rare, life-limiting inherited metabolic disease characterised by accumulation of excess glycine in the body. The disease becomes apparent soon after birth with lethargy, breathing difficulties and neurological symptoms, including seizures. Affected infants suffer epilepsy and profound delay in development and NKH is often fatal in early childhood.
NKH is an autosomal recessive disease, in which one copy of the affected gene is inherited from each parent. Classic NKH is caused by mutations in GLDC or AMT, genes that encode components of the glycine cleavage system). The majority of children carry mutations in GLDC (glycine decarboxylase) and a broad spectrum of mutations have been identified.
The Glycine Cleavage System (GCS) regulates the abundance of glycine by breaking it down in a reaction localised in mitochondria (Figure 1). Glycine is the smallest amino acid and functions both as a neurotransmitter in the central nervous system and as a precursor in a number of biosynthetic processes including production of proteins and purines (for DNA synthesis). The harmful effects of excess glycine in NKH are thought to result, at least in part, through the action at NMDA and glycine receptors in the CNS.
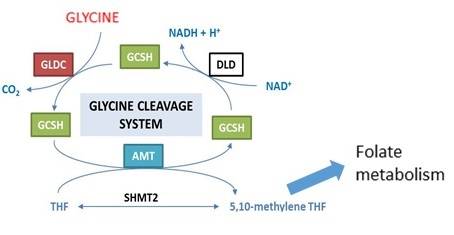
Current treatment for NKH is based on use of sodium benzoate, to lower glycine levels in the body, and management of epilepsy. These treatments do not provide a cure.
Our research aims to better understand the disease process and to develop novel therapies. We using a number of experimental models:
Mouse models of NKH We are currently studying three mouse models that have different mutations in Gldc. This results in 3 main types of abnormality: (i) early defects in pre-natal development of the brain, called termed neural tube defects (NTDs) (note that mutations in GLDC and AMT are also found in some human NTD cases); (ii) hydrocephalus – enlargement of the brain ventricles due to increased pressure of cerebrospinal fluid. Some children with NKH also develop hydrocephalus; (iii) features of NKH [see references for more details].
- GldcGT1
- Mice carry a ‘gene-trap’ construct that prevents the normal transcription of the Gldc gene.
- GldcGT1/GT1 mice that are homozygous for the gene-trap have a 90% lower amount of Gldc gene expression and Gldc protein is undetectable.
- Some embryos (~20%) develop NTDs or hydrocephalus (30%). Remaining mice provide a model of NKH.
- Glycine cleavage system activity (measured in liver) is almost completely absent and these mice provide a model for NKH with higher levels of glycine in blood, urine and body tissues. About half the mice die by 3 months of age.
- The ‘gene-trap’ can be switched on and off by genetic recombination so that the function in different tissues can be investigated separately.
- GldcGT2
- The GldcGT2 mice carry a different gene-trap construct which completely prevents Gldc gene expression.
- This is a more severe model and 60% of GldcGT2/GT2 embryos develop neural tube defects. Few mice survive after birth.
- This model is used to analyse the biochemical and metabolic effects of knockout of the glycine cleavage system.
We also analyse mice carrying one copy of each gene-trap construct (GldcGT1/GT2). They have an intermediate frequency of neural tube defects.
- Gldc missense mutation
- A novel mouse strain has been generated (by CRISPR/Cas targeting) which carries a missense mutation in the DNA which changes one amino acid of the Gldc protein.
- This mutation is identical to a known disease-causing mutation found in a child with NKH (and also in a baby with a neural tube defect).
Other experimental models Alongside work with mouse models we are using:
Cellular models (cell lines that have mutations in GLDC) to study the effect of GCS mutations in human cells. Cell lines can be grown in culture (in a dish in the laboratory) and include:
- Established cell lines, corresponding to cell types that express GLDC, in which we have created GLDC mutations by CRISPR/Cas targeting.
- Induced pluripotent stem cells (iPS cells) generated using skin cells from NKH patients. These iPS cells can now be differentiated to the cell types that are affected in NKH.
Simple organisms. We are using several models in which GCS gene function is deleted in simple organisms which are amenable to high-throughput screening of potential therapeutics and testing of metabolic abnormalities.
Current studies
In the various NKH model systems we are:
- Using mass spectrometryand other biochemical assays to determine the metabolic effects of GLDC mutation in different tissues and cells at various stages of the disease.
- Analysing the effects of GCS abnormalities on cellular functions.
- Investigating neurological effects in the NKH mouse model. e.g. by carrying out EEG recording.
- Asking how brain structure and cellular composition is affected by GLDC mutation.
- Using conditional genetic rescue to examine the requirement for GLDC activity in specific tissues.
This work is aimed at informing development of new treatments and to define therapeutic end-points to test whether such treatments are effective.
Developing novel treatments
- Evaluating small molecules treatment for reduction of glycine, for example as possible alternatives to sodium benzoate.
- Asking whether normalisation of folate metabolism is beneficial. We showed that the one-carbon donor, formate, prevents early defects of brain development raising the question of whether other one-carbon donors may be used at post-natal stages.
- Developing novel gene therapy for NKH aimed at inserting a functioning GLDC gene into the patient cells. These methods are currently being tested in mouse NKH models using (i) AAV vectors to target the brain and (ii) lentiviral vectors to target the liver.
Research Group & Collaborators:
Members of our group working on the GCS and NKH projects include Dr Kit-Yi Leung, Chloe Santos, Dr Sandra Castro, Diana Gold-Diaz, Dr Shabnam Ghazi-Noori.
Dr John Counsell co-leads work on lentiviral gene therapy.
Current work is in close collaboration with Prof. Simon Waddington (UCL Institute for Women’s Health), Dr Filipe Cabreiro (Imperial College London), Prof. Stephanie Schorge (UCL Institute of Pharmacy), Dr Rob Wykes (UCL Institute of Neurology).
