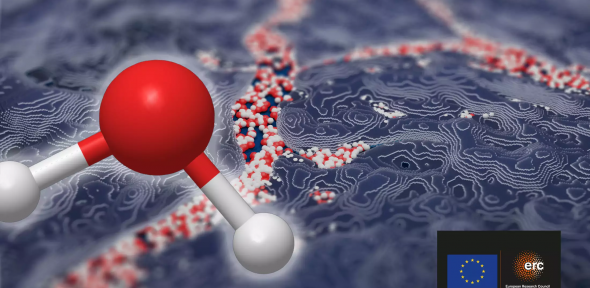
The ICE Group
27/01/2024
17/01/2024
01/12/2023
09/10/2023
04/10/2023
03/09/2023
07/07/2023
27/06/2023 - 30/06/2023
25/06/2023
07/06/2023
25/05/2023
22/04/2023
18/04/2023
30/01/2023
20/01/2023
13/12/2022
12/12/2022
11/12/2022
02/12/2022
25/10/2022
19/10/2022
20/09/2022
25/08/2022
26/07/2022
11/07/2022
15/06/2022
13/06/2022
24/05/2022
10/05/2022
03/05/2022
01/04/2022
10/12/2021
29/11/2021
01/10/2021
26/07/2021
18/07/2021
30/04/2021
9th Nov 2020
7th Oct 2020
13th July 2020
21st May 2020