Close
Close

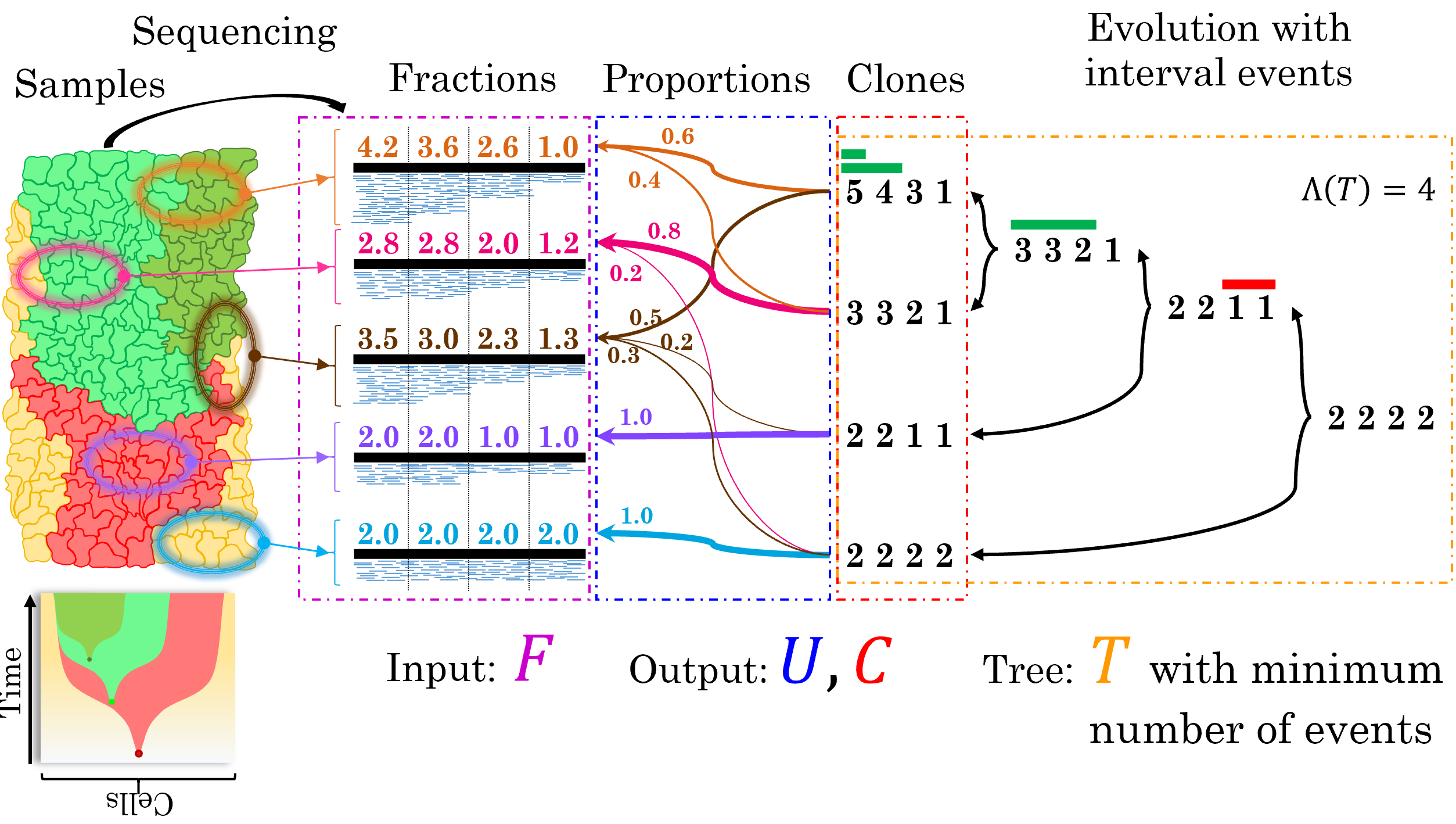
We analyse sequencing data from multiple bulk tumour samples to investigate intra-tumour heterogeneity.
Cancer arises from an evolutionary process where different kinds of somatic mutations are accumulated in the genome of different subpopulations of cells. Such process yields heterogeneous tumours, where distinct subpopulation of cells, or clones, have different complements of somatic mutations. As different tumour clones may have different behaviours, for example different responses to treatment, the identification of somatic mutations from DNA sequencing data have a critical impact on cancer prognosis and diagnosis.
Most cancer sequencing studies perform DNA sequencing of one or more bulk tumour samples, obtained from multiple regions of a primary tumor, matched primary and metastases, or longitudinal samples. However, the inference of somatic mutations from DNA sequencing data of bulk tumour samples is challenging because each samples is a mixture of thousands to millions of different cells from different tumour clones. In such mixtures the signal from the observed sequencing reads is a superposition of the signals from normal cells and distinct tumor clones, which share the same clonal mutations but are distinguished by different subclonal mutations.
Our lab focuses on the design and development of computational methods to deconvolve, or separate, this mixed signal into the individual components arising from each of these clones. Moreover, we design algorithms to investigate spatial heterogeneity from multiple bulk tumour samples and reconstruct tumour phylogenetic trees that describe the history of tumour evolution.