 Close
Close

SHOC of the new: Targeting SHOC2 in RAS mutant cancers
7 August 2019
UCL-led research has identified a promising target that could open up new ways to tackle some of the most common cancers.
More than a third of human cancers have mutations in RAS genes. In addition, mutations in many other genes drive cancerous cell proliferation by disrupting RAS signalling – for example, around three-quarters of non-small cell lung carcinoma, the leading cause of cancer mortality globally, have mutations that affect RAS signalling.
Unfortunately, it has proven very difficult to target RAS directly therapeutically. Instead, researchers have focused on other points in the RAS signalling pathway, particularly the RAF-MEK-ERK kinase cascade [Fig. 1A]. However, RAF inhibitors are not used to treat cancers with mutant RAS genes, because they cause RAF to form dimers that lead to paradoxical activation of ERK. MEK inhibitors are also of limited value in RAS mutant cancers, owing to the rapid emergence of resistance and significant on-target toxicity – MEK is also required for normal cell function [Fig. 1B].
The reliance of many cancers on RAF-MEK-ERK signalling is therefore a potential vulnerability that has been frustratingly difficult to target. To address this issue, CRUK-funded PhD student Greg Jones and colleagues in Pablo Rodriguez-Viciana’s lab have worked with colleagues at Imperial and the Crick Institute on an alternative strategy.
Although much drug development has focused on kinases (such as RAF and MEK), the phosphatases that remove phosphate groups also play a key role in the regulation of signalling pathways. Notably, removal of a specific phosphate group on RAF is a critical step in the formation of ERK-activating RAF dimers. This dephosphorylation relies on a multi-subunit phosphatase, known as the SHOC2 phosphatase complex. Inhibiting SHOC2, the researchers hypothesised, could therefore be an alternative way to indirectly inhibit RAF and dial down ERK activity.
To test this idea, Jones et al. genetically modified mice to eliminate SHOC2. Excitingly, this suppressed tumour development and enhanced survival in a mouse model of mutant RAS lung cancer. Knockdown of SHOC2 also inhibited the proliferation of a range of human RAS mutant cancer cell lines.
Furthermore, SHOC2 elimination had further benefits when combined with MEK inhibitors. For example, it prevented RAF dimerisation-driven ERK activation when MEK inhibitors are used in RAS mutant cancers. It also reduced the risk of resistance – instead of entering a quiescent state, where they are vulnerable to the acquisition of new mutations, cancer cells lacking SHOC2 activated cell death pathways when treated with MEK inhibitors [Fig. 1C].
A final major discovery was that loss of SHOC2 had little impact on mouse cells in culture or adult mice – suggesting that inhibition of SHOC2, unlike MEK targeting, would have little impact on normal cell function.
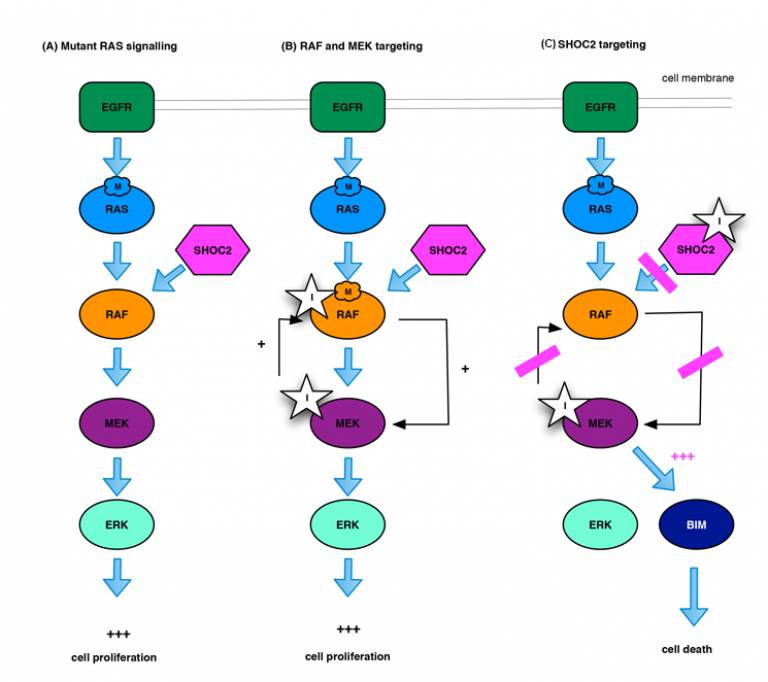
(A) Mutant RAS signalling: Signalling through the RAF–MEK–ERK kinase cascade drives cell proliferation.
(B) MEK targeting: In RAS mutant cells, RAF targeting leads to dimerisation of RAF and activation of MEK and ERK, while inhibition of MEK disrupts feedback loops, similarly reactivating RAF dimerisation and ERK signalling.
(C) SHOC2 targeting: SHOC2 inhibition prevents MEK inhibitor-induced RAF dimerisation, blocking signalling through the RAF–MEK–ERK kinase cascade, and combines with MEK inhibition to drive cell death.
This conclusion is backed up by the results of a related study from CRUK-funded PhD student Isabel Boned del Rio and colleagues in Pablo Rodriguez-Viciana’s lab. Looking in detail at the cellular role of SHOC2, the group identified both SHOC2-dependent and -independent mechanisms of ERK activation. Importantly, aberrant signalling in RAS mutant cancer cells is entirely dependent on the SHOC2-dependent pathway. This suggests that, in contrast to targeting RAF/MEK/ERK core pathway components that inhibit global ERK signaling, blocking SHOC2 function would interfere with oncogene-driven ERK activation, while not affecting other aspects of normal ERK regulation mediated through SHOC2-independent mechanisms [Fig. 2].
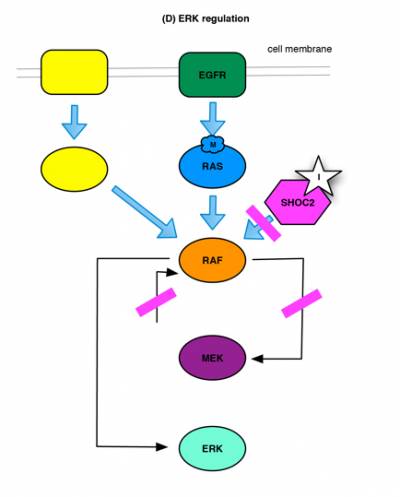
(D) ERK regulation: ERK is regulated through multiple signalling pathways. Inhibition of SHOC2 blocks oncogene-driven ERK activation without interfering with other regulatory pathways.
Together, the studies therefore suggest that targeting SHOC2 – on its own or to augment the action of MEK inhibitors – could be an effective strategy to achieve better therapeutic margins against RAS-driven tumours. Furthermore, it could provide new options not just for RAS mutant cancers – including a very high proportion of pancreatic cancers – but also for other cancers with mutations affecting RAS signalling, such as EGFR mutations, found in one in ten lung cancers.
“Strikingly, in RAS mutant cells, most resistance mechanisms lead to ERK-pathway reactivation, highlighting the strong ‘oncogene addiction’ of these cancers to ERK signalling. This could be their Achilles heel. We’ve shown that inhibiting SHOC2 could be a way of exploiting this vulnerability, opening the door to new treatments for the world’s most common cancers.” Greg Jones, PhD student,UCL Cancer Institute.
“On-target toxicity has limited the usefulness of ERK pathway inhibitors. Our work suggests that targeting SHOC2 could prevent oncogene-driven ERK activation but still leave ERK free to perform its normal cellular functions.” Isabel Boned del Rio, PhD student UCL Cancer Institute.
Further information
- Research paper: SHOC2 phosphatase-dependent RAF dimerization mediates resistance to MEK inhibition in RAS-mutant cancers. Nature Communications.
- Reseach paper: SHOC2 complex-driven RAF dimerization selectively contributes to ERK pathway dynamics
- Dr Pablo Rodriguez-Viciana academic profile
- Lead image: Pancreatic cancer cells. National Cancer Institute \ USC Norris Comprehensive Cancer Center