
| |
Neal Skipper (CMMP & LCN)
 |
- Liquids, Complex & Confined Fluids
- Clay Minerals & Layered Double Hydroxides
- Graphite Intercalates & Carbon Nanostructures
- Neutron & X-ray Scattering
- Computer Modelling & Statistical Physics
- Hydrogen Storage & Energy Materials
|
|


|
Neal Skipper is Professor of Physics and a founder member of the Condensed Matter and Materials Physics group
(CMMP) at University College London. He is also a Principal Investigator
in the London Centre for Nanotechnology (LCN).
Research Interests
Our research aims to understand and control the properties of materials, by building up a picture of where the atoms are what the atoms do. This approach also allows us to design new materials that will perform particular functions for us. For example, we are currently creating solids that will be able to store hydrogen for the supply of clean energy, and we are devising ways to process nanotubes and graphene so that we can exploit the remarkable properties of these forms of carbon. We also have long-standing interests in the more fundamental properties of liquids and non-crystalline materials. We are experts in the handling of liquid ammonia and metal-ammonia solutions, and are therefore able to exploit its extraordinary properties as a solvent. These include its ability to dissolve very large concentrations of electrons, giving us unique metallic liquids of very low density. We are also very interested in the way that aromatic molecules interact with each other via their π-electrons. These interactions are a key factor in many important biological and chemical processes.
We use a variety of experimental and computational techniques in our research. In addition to our laboratories at UCL, we also make extensive use of international neutron and X-ray scattering facilities. For example the
ISIS Neutron Source at the Rutherford Appleton Laboratory, and the Institute Laue-Langevin in Grenoble.
Other Activities
I was Co-Founder of Cella Energy Ltd which is a company specialising in hydrogen storage materials, spun-out from the Rutherford Appleton Laboratory, UCL and the University of Oxford. Cella Energy has developed a method using a low-cost process called coaxial electrospinning or electrospraying. This traps a complex chemical hydride inside a nano-porous polymer, speeds up the kinetics of hydrogen desorption, reduces the temperature at which the desorption occurs and filters out many if not all of the damaging chemicals. It also protects the hydrides from oxygen and water, making it possible to handle it in air. Cella Energy is winner of the: Shell Springboard Award, Energy Innovation Award, and Energy Storage Challenge.
Some Recent Publications
| Strong structuring arising from weak cooperative O-H···π and C-H···O hydrogen bonding in benzene-methanol solution Di Mino, C.; Seel, A.G.;, Clancy, A.J.; Headen, T.F.; Földes, T.; Rosta, E.; Sella, A.; Skipper, N.T. Nature Communications 14, 5900 (2023).
DOI: https://doi.org/10.1038/s41467-023-41451-y |
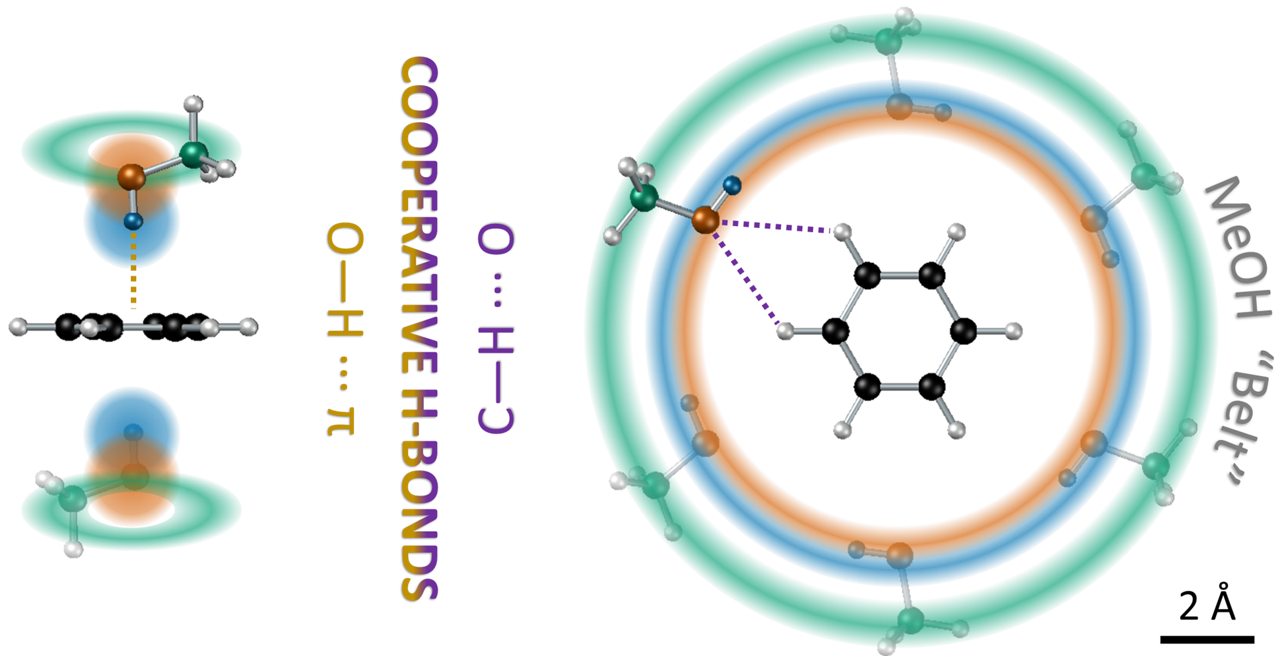 |
Weak hydrogen bonds, such as O-H···π and C-H···O, are thought to direct biochemical assembly, molecular recognition, and chemical selectivity but are seldom observed in solution. We have used neutron diffraction combined with H/D isotopic substitution to obtain a detailed spatial and orientational picture of the structure of benzene-methanol mixtures. Our analysis reveals that methanol fully solvates and surrounds each benzene molecule. The expected O-H···π interaction is highly localised and directional, with the methanol hydroxyl bond aligned normal to the aromatic plane and the hydrogen at a distance of 2.30Å from the ring centroid. Simultaneously, the tendency of methanol to form chain and cyclic motifs in the bulk liquid is manifest in a highly templated solvation structure in the plane of the ring. The methanol molecules surround the benzene so that the O-H bonds are coplanar with the aromatic ring while the oxygens interact with C-H groups through simultaneous bifurcated hydrogen bonds. This demonstrates that weak hydrogen bonding can modulate existing stronger interactions to give rise to highly ordered cooperative structural motifs that persist in the liquid phase. |
| Production of phosphorene nanoribbons Watts, M.C.; L Picco, L.; Russell-Pavier, F.S.; Cullen, P.L.; Miller, T.S.; Bartus, S.F.; Payton, O.D.; Skipper, N.T.; Tileli, V.; Howard, C.A.; Nature 568, 216-220 (2019).
DOI: https://doi.org/10.1038/s41586-019-1074-x |
 |
Here we present a method for creating quantities of high-quality, individual PNRs by ionic scissoring of macroscopic black phosphorus crystals. This top-down process results in stable liquid dispersions of PNRs with typical widths of 4-50 nm, predominantly single-layer thickness, measured lengths of up to 75 ?m and aspect ratios of up to 1,000. The nanoribbons are atomically flat single crystals, aligned exclusively in the zigzag crystallographic orientation. The ribbons have remarkably uniform widths along their entire lengths, and are extremely flexible. These properties-together with the ease of downstream manipulation via liquid-phase methods-should enable the search for predicted exotic states and an array of applications in which PNRs have been predicted to offer transformative advantages. These applications range from thermoelectric devices to high-capacity fast-charging batteries and integrated high-speed electronic circuits. |
| Formation of Methane Hydrate in the Presence of Natural and Synthetic Nanoparticles Cox, S.J.; Taylor, D.J.F.; Youngs, T.G.A.; Soper, A.K.; Totton, T.S.; Chapman, R.G.; Arjmandi, M.; Hodges, M.G.; Skipper, N.T.; Michaelides.A. J. Am, Chem. Soc.
140, 3277-3284 (2018).
DOI: 10.1021/jacs.7b12050 |
 |
Natural gas hydrates occur widely on the ocean-bed and in permafrost regions, and have potential as an untapped energy resource. Their formation and growth, however, poses major problems for the energy sector due to their tendency to block oil and gas pipelines, whereas their melting is viewed as a potential contributor to climate change. Although recent advances have been made in understanding bulk methane hydrate formation, the effect of impurity particles, which are always present under conditions relevant to industry and the environment, remains an open question. Here we present results from neutron scattering experiments and molecular dynamics simulations that show that the formation of methane hydrate is insensitive to the addition of a wide range of impurity particles. Our analysis shows that this is due to the different chemical natures of methane and water, with methane generally excluded from the volume surrounding the nanoparticles. This has important consequences for our understanding of the mechanism of hydrate nucleation and the design of new inhibitor molecules. |
| Charged Carbon Nanomaterials: Redox Chemistries of Fullerenes, Carbon Nanotubes, and Graphenes Clancy, A.J.; Bayazit, M.K.; Hodge, S.A.; Skipper, N.T.; Howard, C.A.; Shaffer, M.S.P. Chemical Reviews
118, 7363-7408 (2018).
DOI: 10.1021/acs.chemrev.8b00128 |
 |
Since the discovery of buckminsterfullerene over 30 years ago, sp2-hybridised carbon nanomaterials (including fullerenes, carbon nanotubes, and graphene) have stimulated new science and technology across a huge range of fields. Despite the impressive intrinsic properties, challenges in processing and chemical modification continue to hinder applications. Charged carbon nanomaterials (CCNs), formed via the reduction or oxidation of these carbon nanomaterials, facilitate dissolution, purification, separation, chemical modification, and assembly. This approach provides a compelling alternative to traditional damaging and restrictive liquid phase exfoliation routes. The broad chemistry of CCNs not only provides a versatile and potent means to modify the properties of the parent nanomaterial but also raises interesting scientific issues. This review focuses on the fundamental structural forms: buckminsterfullerene, single-walled carbon nanotubes, and single-layer graphene, describing the generation of their respective charged nanocarbon species, their interactions with solvents, chemical reactivity, specific (opto)electronic properties, and emerging applications. |
| Ionic solutions of two-dimensional materials. Cullen, P.L.; Cox, K.M.; Bin Subhan, M.K.; Picco, L.; Payton, O.D.; Buckley, D.J.; Miller, T.S.; Hodge, S.A.; Skipper, N.T.; Tileli, V.; Howard. C.A. Nature Chemistry
9, 244-249 (2017).
DOI: https://doi.org/10.1038/nchem.2650 |
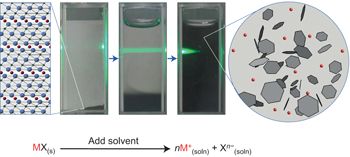 |
Strategies for forming liquid dispersions of nanomaterials typically focus on retarding reaggregation, for example via surface modification, as opposed to promoting the thermodynamically driven dissolution common for molecule-sized species. Here we demonstrate the true dissolution of a wide range of important 2D nanomaterials by forming layered material salts that spontaneously dissolve in polar solvents yielding ionic solutions. The benign dissolution advantageously maintains the morphology of the starting material, is stable against reaggregation and can achieve solutions containing exclusively individualized monolayers. Importantly, the charge on the anionic nanosheet solutes is reversible, enables targeted deposition over large areas via electroplating and can initiate novel self-assembly upon drying. Our findings thus reveal a unique solution-like behaviour for 2D materials that enables their scalable production and controlled manipulation. |
| Trajectory of the Selective Dissolution of Charged Single-Walled Carbon Nanotubes. Buckley, D.J.; Hodge, S.A.; De Marco, M.; Hu, S.; Anthony, D.B.; Cullen, P.Ll; Skipper, N.T.; Shaffer, M.S.P.; Howard, C.A. J. Phys. Chem. C 2017, 121, 21703-21712 (2017).
DOI: 10.1021/acs.jpcc.7b06553 |
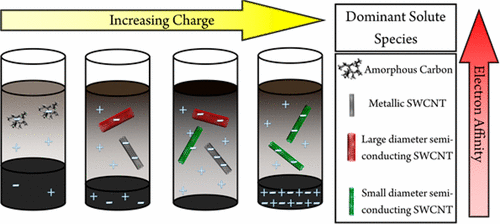 |
Single-walled carbon nanotubes (SWCNTs) are typically produced as a mixture of different lengths and electronic types. Methods for sorting these as-produced mixtures typically require damaging and unscalable techniques to first overcome the strong bundling forces between the nanotubes. Previously, it has been shown that negatively charging SWCNTs can lead to their thermodynamically driven, gentle dissolution in polar solvents, and moreover that this process can selectively dissolve different SWCNT species. However, there are several conflicting claims of selectivity that must be resolved before the full potential of this method for scalably postprocessing SWCNTs can be realized. Here we carefully investigate dissolution as a function of charge added to the as-produced SWCNT sample, using a range of complementary techniques. We uncover a far richer dependence on charge of SWCNT dissolution than previously imagined. At low values of charge added, amorphous carbons preferentially dissolve, followed sequentially by metallic, larger diameter (>9 A) semiconducting SWCNTs, and finally smaller diameter semiconducting SWCNTs as more charge is added. At an optimal stoichiometry of NaC10, the dissolution yield is maximized across all species. However, at higher charge the larger diameter and metallic SWCNTs are so highly charged that they are no longer soluble, leaving smaller diameter SWCNTs in solution. Our results clearly demonstrate two interconnected mechanisms for dissolution: the sequential charging of the SWCNTs and their solution thermodynamics. This work reconciles conflicting reports in the literature, demonstrates that upon charge added the different SWCNTs behave like discrete molecular species, and points toward selective dissolution as a scalable method for SWCNT separation. |
| Electron Solvation and the Unique Liquid Structure of a Mixed-Amine Expanded Metal: The Saturated Li-NH3-MeNH2 System Seel, A.G.;, Swan, H.; Bowron, D.T.; Wasse, J.C.; Weller, T.; Edwards, P.P.; Howard, C.A.; Skipper, N.T.; Angewandte Chemie International Edition
129, 1583-1587 (2017).
DOI: https://doi.org/10.1002/ange.201609192 |
 |
Metal-amine solutions provide a unique arena in which to study electrons in solution, and to tune the electron density from the extremes of electrolytic through to true metallic behavior. The existence and structure of a new class of concentrated metal-amine liquid, Li-NH3-MeNH2, is presented in which the mixed solvent produces a novel type of electron solvation and delocalization that is fundamentally different from either of the constituent systems. NMR, ESR, and neutron diffraction allow the environment of the solvated electron and liquid structure to be precisely interrogated. Unexpectedly it was found that the solution is truly homogeneous and metallic. Equally surprising was the observation of strong longer-range order in this mixed solvent system. This is despite the heterogeneity of the cation solvation, and it is concluded that the solvated electron itself acts as a structural template. This is a quite remarkable observation, given that the liquid is metallic. |
| Structure and Dynamics of Molecular Hydrogen in the Interlayer Pores of a Swelling 2:1 Clay by Neutron Scattering. Edge, J. E.; Skipper, N. T.; Fernandez-Alonso, F.; Lovell, A.; Gadipelli, S.; Bennington, S.M.; Garcia Sakai, V.; Youngs, T. G. A. J. Phys. Chem. C
118, 25740-25747 (2014).
DOI: 10.1021/jp5082356 |
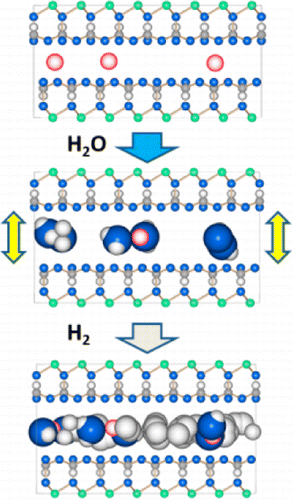 |
Neutron scattering has been used to reveal the structure and dynamics of molecular H2 physisorbed into the two-dimensional pores of sparingly hydrated Ca-laponite clay. Thermal pretreatment of the clay at 415 K under vacuum yielded an interlayer composition in the 1.0-1.5 water molecules per Ca2+ cation range and provided a vacant gallery height of 2.82 A. This value is very well matched to the diameter of molecular hydrogen and allows intercalation of H2 up to the point where a liquid-like monolayer is formed within the clay. At a low coverage of 0.1 H2 per cation the isosteric heat of adsorption is 9.2 kJmol-1. Quasielastic neutron scattering experiments conducted at 40-100 K reveal two populations of H2 within the clay. First, we find molecules that are localized close to the partially hydrated Ca2+ cations. Second, we identify a more mobile liquid-like population whose motion is captured by jump diffusion. At 40 K, the H2 diffusion coefficient is 2.3 +/- 0.5 x 10-5 cm2s-1. This is an order of magnitude slower than the value extrapolated from bulk liquid H2. |
| Electrochemical Processing of Discrete Single-Walled Carbon Nanotube Anions. Hodge, S.M.; Fogden, S.; Howard, C.A.; Skipper, N.T.; Shaffer, M.S.P.; ACS Nano
7, 1769-1778 (2013).
DOI: 10.1021/nn305919p |
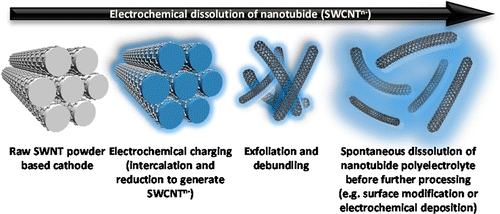 |
The dissolution of single-walled carbon nanotubes (SWCNTs) remains a fundamental challenge, reliant on aggressive chemistry or ultrasonication and lengthy ultracentrifugation. In contrast, simple nonaqueous electrochemical reduction leads to spontaneous dissolution of individualized SWCNTs from raw, unprocessed powders. The intrinsic electrochemical stability and conductivity of these nanomaterials allow their electrochemical dissolution from a pure SWCNT cathode to form solutions of individually separate and distinct (i.e., discrete) nanotube anions with varying charge density. The integrity of the SWCNT sp2 framework during the charge/discharge process is demonstrated by optical spectroscopy data. Other than a reversible change in redox/solvation state, there is no obvious chemical functionalization of the structure, suggesting an analogy to conventional atomic electrochemical dissolution. The heterogeneity of as-synthesized SWCNT samples leads to the sequential dissolution of distinct fractions over time, with fine control over the electrochemical potential. Initial preferential dissolution of defective nanotubes and carbonaceous debris provides a simple, nondestructive means to purify raw materials without recourse to the usual, damaging, competitive oxidation reactions. Neutral SWCNTs can be recovered either by electroplating at an anode or by reaction with a suitable electrophile. |
| Structure and Morphology of Charged Graphene Platelets in Solution by Small-Angle Neutron Scattering. Milner, E.M.; Skipper, N. T.; Howard, C.A.; Shaffer, M.S.P.; Buckley, D.J.; Rahnejat, K.A.; Cullen; P.L.; Heenan, R.K.; Lindner, P.; Schweins, R. J. Am Chem. Soc.
134, 8302-8305 (2012).
DOI: 10.1021/ja211869u |
 |
Solutions of negatively charged graphene (graphenide) platelets were produced by intercalation of nanographite with liquid potassium-ammonia followed by dissolution in tetrahydrofuran. The structure and morphology of these solutions were then investigated by small-angle neutron scattering. We found that >95 vol % of the solute is present as single-layer graphene sheets. These charged sheets are flat over a length scale of >150 A in solution and are strongly solvated by a shell of solvent molecules. Atomic force microscopy on drop-coated thin films corroborated the presence of monolayer graphene sheets. Our dissolution method thus offers a significant increase in the monodispersity achievable in graphene solutions. |
| Scalable Method for the Reductive Dissolution, Purification, and Separation of Single-Walled Carbon Nanotubes Fogden, S; Howard, C.A.; Heenan, R.K.; Skipper, N.T.; Shaffer, M.S.P. ACS Nano 6, 54-62 (2012).
DOI: 10.1021/nn2041494 |
 |
As synthesized, bulk single-walled carbon nanotube (SWNT) samples are typically highly agglomerated and heterogeneous. However, their most promising applications require the isolation of individualized, purified nanotubes, often with specific optoelectronic characteristics. A wide range of dispersion and separation techniques have been developed, but the use of sonication or ultracentrifugation imposes severe limits on scalability and may introduce damage. Here, we demonstrate a new, intrinsically scalable method for SWNT dispersion and separation, using reductive treatment in sodium metal-ammonia solutions, optionally followed by selective dissolution in a polar aprotic organic solvent. In situ small-angle neutron scattering demonstrates the presence of dissolved, unbundled SWNTs in solution, at concentrations reaching at least 2 mg/mL; the ability to isolate individual nanotubes is confirmed by atomic force microscopy. Spectroscopy data suggest that the soluble fraction contains predominately large metallic nanotubes; a potential new mechanism for nanotube separation is proposed. In addition, the G/D ratios observed during the dissolution sequence, as a function of metal:carbon ratio, demonstrate a new purification method for removing carbonaceous impurities from pristine SWNTs, which avoids traditional, damaging, competitive oxidation reactions. |
| Activation and local structural stability during the thermal decomposition of Mg/Al-hydrotalcite by total neutron scattering. Mourad, M.C.D.; Mokhtar, M.; Tucker, M.G.; Barney, E.R.; Smith, R.I.; Alyoubi, A.O.; Basahel, S.N.; Shaffer M.S.P.; Skipper, N.T.J. Mater. Chem. 21, 15479-15485 (2011).
DOI: 10.1039/C1JM11530H |
 |
The activation of synthetic hydrotalcite, the carbonated layered double hydroxide (LDH) with ratio Mg2+/Al3+ = 3 and structural formula [Mg6Al2(OH)16]2+ CO32: n(H2O), has been investigated using neutron and X-ray diffraction. In situ neutron diffraction was used to follow the structural phase transformations during the thermal decomposition (calcination) of hydrotalcite under vacuum in the temperature range 298-723 K, and during which the residual gas evolved by the sample was analysed by mass spectrometry. Detailed structural information of the LDH and mixed metal oxides (MMOs) was extracted from both the Bragg peaks and the total scattering. These two analysis techniques provide complementary insight into the relevant transition mechanisms, since Bragg diffraction originates from long-range periodicities within the samples while total scattering reveals the subtleties of the local atomic environment. This latter information is particularly important for our understanding of catalytic activity since it elucidates the local metal coordination. We find that, during the calcination process, the local environment around the metal centres is robust, as the various stages during the phase transition have identical local structures. The implications of these new results for the nature of the MMOs is discussed in relation to the well-studied, reverse, rehydration reaction, and the high propensity of trivalent Al ions to migrate to tetrahedrally-coordinated lattice sites. |
| Chiral interactions of histidine in a hydrated vermiculite clay. Fraser, DG; Greenwell, HC; Skipper, NT; Smalley, MV; Wilkinson, MA; Deme, B; Heenan, RK. Phys. Chem. Chem. Phys. 13, 825-830 (2011).
DOI: 10.1039/c0cp01387k |
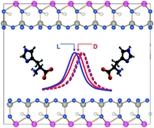 |
Recent work shows a correlation between chiral asymmetry in non-terrestrial amino acids extracted from the Murchison meteorite and the presence of hydrous mineral phases in the meteorite [D. P. Glavin and J. P. Dworkin, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5487-5492]. This highlights the need for sensitive experimental tests of the interactions of amino acids with clay minerals together with high level computational work. We present here the results of in situ neutron scattering experiments designed to follow amino acid adsorption on an exchanged, 1-dimensionally ordered n-propyl ammonium vermiculite clay. The vermiculite gel has a (001) d-spacing of order 5 nm at the temperature and concentration of the experiments and the d-spacing responds sensitively to changes in concentration, temperature and electronic environment. The data show that isothermal addition of D-histidine or L-histidine solutions of the same concentration leads to an anti-osmotic swelling, and shifts in the d-spacing that are different for each enantiomer. This chiral specificity, measured in situ, in real time in the neutron beam, is of interest for the question of whether clays could have played an important role in the origin of biohomochirality. |
| The structure of π-π interactions in aromatic liquids. Headen TF, Howard CA, Skipper NT, Wilkinson MA, Bowron DT, Soper AK. J. Am. Chem. Soc. 132, 5735-5742 (2010).
DOI: 10.1021/jp8083502 JACS cover. |
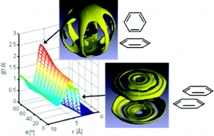 |
High-resolution neutron diffraction has been used in conjunction with hydrogen/deuterium isotopic labeling to determine with unprecedented detail the structure of two archetypal aromatic liquids: benzene and toluene. We discover the nature of aromatic π-π interactions in the liquid state by constructing for the first time a full six-dimensional spatial and orientational picture of these systems. We find that in each case the nearest neighbor coordination shell contains approximately 12 molecules. Benzene is the more structured of the two liquids, showing, for example, a sharper nearest neighbor coordination peak in the radial distribution function. Superficially the first neighbor shells appear isotropic, but our multidimensional analysis shows that the local orientational order in these liquids is much more complex. At small molecular separations (<5 A) there is a preference for parallel π-π contacts in which the molecules are offset to mimic the interlayer structure of graphite. At larger separations (>5 A) the neighboring aromatic rings are predominantly perpendicular, with two H atoms per molecule directed toward the acceptor's π orbitals. The so-called "anti-hydrogen-bond" configuration, proposed as the global minimum for the benzene dimer, occurs only as a saddle point in our data. The observed liquid structures are therefore fundamentally different than those deduced from the molecular dimer energy surfaces. |
| Synthesis of graphene-like nanosheets and their hydrogen adsorption capacity. Srinivas, G.; Zhu, Y.; Piner, R.; Skipper, N.T.; Ellerby, M.; Ruoff, R. Carbon 48, 630-635 (2010).
DOI: 10.1016/j.carbon.2009.10.003 |
 |
Graphene-like nanosheets have been synthesized by the reduction of a colloidal suspension of exfoliated graphite oxide. The morphology and structure of the graphene powder sample was studied using scanning electron microscopy, transmission electron microscopy, X-ray diffraction and Raman spectroscopy. The graphene sheets are found to be in a highly agglomerated state, with many wrinkles. The sample has a BET surface area of 640 m2/g as measured by nitrogen adsorption at 77 K. Hydrogen adsorption-desorption isotherms were measured in the temperature range 77-298 K and at pressures of up to 10 bar. This gives hydrogen adsorption capacities of about 1.2 wt.% and 0.1 wt.% at 77 K and 298 K, respectively. The isosteric heat of adsorption is in the range of 5.9-4 kJ/mol, indicating a favourable interaction between hydrogen and surface of the graphene sheets. The estimated room temperature H2 uptake capacity of 0.72 wt.% at 100 bar and the isosteric heat of adsorption of our sample are comparable to those of high surface area activated carbons, however significantly better than the recently reported values for graphene and a range of other carbon and nanoporous materials; single and multi walled carbon nanotubes, nanofibers, graphites and zeolites. |
| A Solution Selection Model for Coaxial Electrospinning and Its Application to Nanostructured Hydrogen Storage Materials. Kurban, Z.; Lovell, A.; Bennington, S.M.; Jenkins, D.W.K.; Ryan, K.R.; Jones, M.O.; Skipper, N.T.; David, W.I.F. J. Phys. Chem. C 114, 21201-21213 (2010).
DOI: 10.1021/jp107871v |
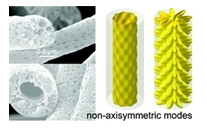 |
Coaxial electrospinning was used to encapsulate the complex hydride ammonia borane in polystyrene to improve its properties as a hydrogen storage material. A solvent selection system was developed by using the Hansen solubility parameters to facilitate the choice of compatible solvents for core and shell. This enabled systematic optimization of the parameters needed for successful coelectrospinning. This approach has general application for any multiphase electrospinning system, including ones where the core is highly conducting or nonpolymeric. The resulting fiber morphologies depend strongly on the degree of miscibility of core and shell solutions. Fibers spun from immiscible core?shell solutions had a classic coaxial structure. Fibers produced from semimiscible core-shell solutions were highly porous, with inclusions extending through the fiber and an ordered radial and longitudinal distribution of nanoscale pores on the fiber surface. We suggest that this type of porosity may be due to an instability created in the nonaxisymmetric modes at the core-shell interface, resulting in intrusion of the core into the shell polymer. These controllably porous structures have numerous potential applications including materials templating or drug delivery. In the porous fibers, the temperature of the first hydrogen release of ammonia borane is reduced to 85 C. This result suggests a nanostructured hydride, but a large mass loss indicates that much of the ammonia borane is expelled on heating. The coaxial fibers, in contrast, appear to encapsulate the hydride successfully. The coaxial and porous fibers alike showed no significant release of borazine, suggesting two different suppression mechanisms for this impurity. |
| Computer Simulations of Fulleride Anions in Metal-Ammonia Solutions. Howard, C.A.; Skipper, N.T. J. Phys. Chem. B 113, , 3324-3332 (2009).
DOI: 10.1021/jp8083502 |
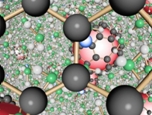 |
Monte Carlo computer simulation has been used to study the energetics and local structure of fulleride anions C60n+ (n = 0, 2, 4, 6) in metal-ammonia solutions. We find that the enthalpy of dissolution is markedly favorable only for n = 2 and 4, which is in line with experimental observations. Analysis of the structure developed around the fulleride anions shows two strong solvation shells of ammonia at distances of around 6.75 and 9.5 A from the fulleride center of mass. This is in excellent agreement with high-resolution neutron diffraction studies of K5C60 in ammonia. The uncharged fullerene (n = 0) induces no discernible orientational order in the neighboring solvent. In contrast to this, there is progressively stronger hydrogen-bonding of the first-shell solvent to the anions (n = 2, 4, 6), approaching one hydrogen-bond per molecule for n = 6. This maximum of one hydrogen bond per ammonia to the fulleride anion is found to permit intersolvent hydrogen bonding within and across the solvation shells similar to that found in bulk liquid ammonia. Comparison of the cations Li+, Na+, K+, and Ca2+ shows that only the potassium has a tendency to form direct ion-pair complexes with the fulleride anion. This work therefore highlights the mechanisms by which metal-ammonia solutions are able to dissolve high concentrations of fullerene salts.td>
|
| Evidence for Asphaltene Nanoaggregation in Toluene and Heptane from Molecular Dynamics Simulations. Headen, T.F.; Boek, E.S.; Skipper, N.T. Energy & Fuels 23 1220-1229 (2009). DOI: 10.1021/ef800872g
| 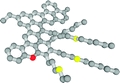 |
Molecular dynamics simulation techniques have been used to study the nanoaggregation of one resin and two asphaltene structures, generated by an updated quantitative molecular representation (QMR) technique (Boek, E. S., Yakovlev, D. S., and Headen, T. F. Energy Fuels, manuscript submitted), in toluene and heptane. Analysis of the simulation trajectories, to yield the separation of asphaltene or resin pairs, over a 20 ns simulation has been used to investigate aggregation dynamics. The structure of aggregates has been investigated by the calculation of the asphaltene-asphaltene and resin?resin radial distribution functions, g(r), and the average angle between the polyaromatic planes as a function of separation. We calculate, for the first time, the asphaltene?asphaltene potential of mean force (PMF) from the g(r) and separately by a constraint force method. In general, it is observed that the asphaltenes form dimers and trimers in both toluene and heptane. Once formed, the dimers and trimers can separate and reform other aggregates with other asphaltene molecules. Aggregates persist for longer in heptane than in toluene. The resin molecule forms no aggregates in toluene, with some aggregation occurring in heptane. Significant peaks in the asphaltene?asphaltene g(r) are seen in both toluene and heptane between 0.5 and 1 nm separation. This is strong evidence for asphaltene nanoaggregation from molecular dynamics simulations. At the lowest separations, the angle between the aromatic planes is close to parallel. The calculated potential of the mean force gives similar results for both methods. For the asphaltene molecules in both toluene and heptane, free energy of dimer formation ranged from 6.6 to 12.1 kJ mol-1. |
| Quantum delocalization of molecular hydrogen in alkali-graphite intercalates. Lovell, A.; Fernandez-Alonso, F.; Skipper, N.T.; Refson, K.; Bennington, S.M.; Parker, S.F. Phys. Rev. Lett. 101, 126101 (2008). DOI: 10.1103/PhysRevLett.101.126101
| 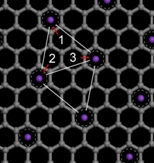 |
The adsorption of molecular hydrogen (H2) in the graphite intercalation compound KC24 is studied both experimentally and theoretically. High-resolution inelastic neutron data show spectral features consistent with a strong pinning of H2 along a single axis. First-principles calculations provide novel insight into the nature of H2 binding in intercalates but fail to account for the symmetry of the H2 orientational potential deduced from experiment. The above discrepancy disappears once the H2 center of mass is allowed to delocalize in the quantum-mechanical sense across three vicinal adsorption sites, naturally leading to the well-known saturation coverage of ~2H2 per metal atom in this material. Our results demonstrate that H2 storage in metal-doped carbon substrates can be severely affected by hitherto unexplored quantum-mechanical effects. |
|  |








 - © Arthur Lovell 2005")

|
