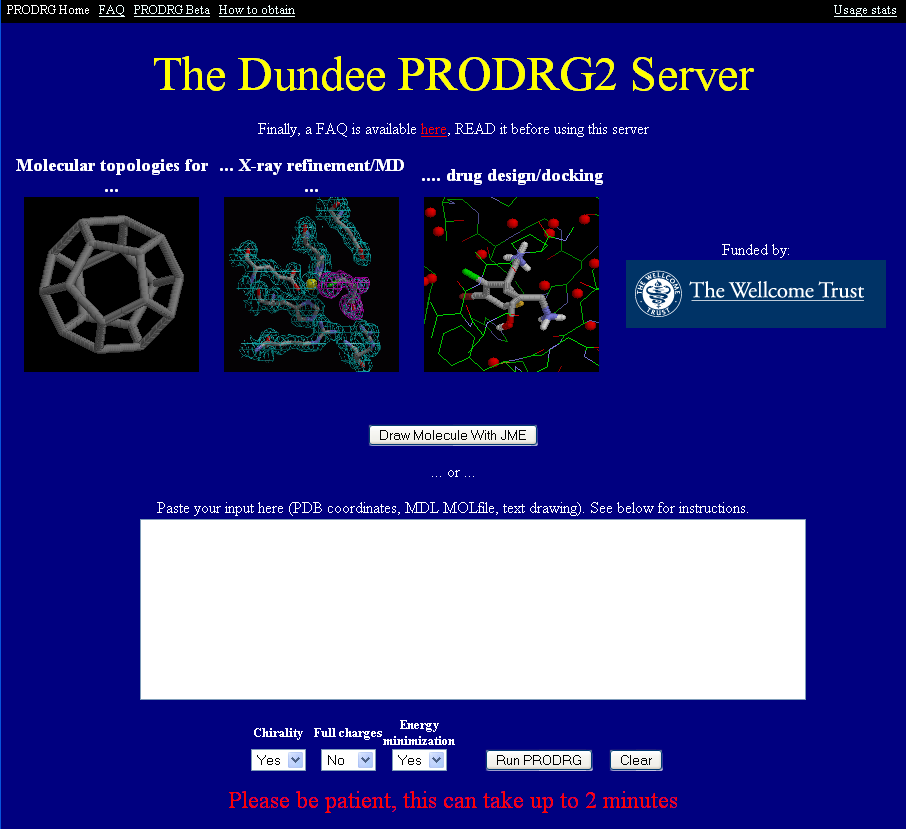
Creating a ligand dictionary
When refining non-protein atoms you will need to generate a pdb file and the appropriate dictionaries for the different refinement programs. You can do this using the excellent online PRODRG2 website. All you have to do is either paste in the pdb coordinates of your ligand (perhaps obtained from an earlier structure) or alternatively click on the "Draw molecule with JME" in the PRODRG2 window:
Once you have your ligand coordinates in the window (sometimes its worth re-numbering the atoms as PRODRG2 uses letters rather than numbers to count carbon atoms) you simply click on "Run PRODRG". The program will regularise your ligand (ie make sure the bond length and angles are OK) and then give you a picture of your ligand along with a variety of dictionary files. Once you have checked if your molecule look OK you can download the various files by simply clicking on the links, highlighting the text and then pasting them into a text editor.
To get the ligands pdb file (to build into your structure using a modelling program such as Coot) click on the "pdb no H's" link.
For refmac you will need to save the file as a .cif file and then select the file in the dictionary section of the Refmac input window.
For CNS you will need to save a .top and a .par file (they are both listed) and then assign the dictionaries in the .inp file.
For shelx you will need to copy the constraints and paste them into the top section of shelxpro.ins
For phenix.refine you can use any of the above (although I normally use the refmac .cif files).