 Close
Close

UCL Genetics Institute
Welcome
Our research is both fundamental and applied, and we believe there does not need to be a divide between the two. Important Science is by definition fundamental, and is also likely to have downstream applications.
We develop computational tools and apply them to large genetic datasets to address questions ranging from the origin and evolution of anatomically modern humans, the genetic determinants of various human diseases, improvement of crop varieties to antibiotic resistance in bacteria.

About Us


News
Research themes

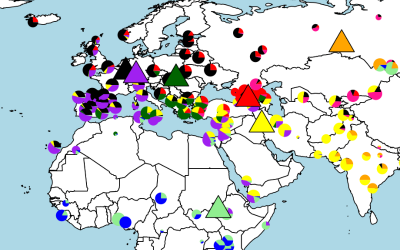

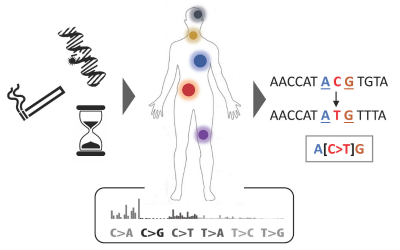


Study
UGI staff teach on various undergraduate programs, including the Genetics pathway that is available for BSc and MSci degrees in Biological Sciences.
Undergraduates

Masters level training is offered through our MSc in Human Genetics Disease. Our PhD students are funded through a diverse range of programmes.
Masters and PhDs

Our growing and vibrant research community is always looking for the brightest minds - see current opportunities from fellowships, PhDs and more.


Read the Paper

Read the Paper

Read the Paper

Read the Paper

Read the Paper

Read the Paper
