CSAR GRANT GR/N20607/01 FINAL REPORT
MATERIALS CHEMISTRY CONSORTIUM
27 March 2004
Introduction
High capability computing resources exploiting massive parallelism provided by CSAR facilities have been used, by our consortium, in wide ranging studies of structures, properties and reactivity of inorganic solids and nanoparticles. We have employed large-scale, static and dynamical simulations to elucidate structural and dynamical properties of complex materials and to determine reaction mechanisms within solids and at surfaces. Our studies have exploited state-of-the-art quantum mechanical techniques based upon density functional theory (DFT), Hartree-Fock, advanced classical interatomic potential simulations and hybrid QM/MM methods.
Our science has been grouped around the three themes of A) structure and transport, B) surface science and reactivity, C) crystal growth and nucleation. Highlights of our work include:
· Identification of a novel class of low dosage inhibitor for clathrate hydrate formation [1]; subsequent development of the new lead compound has led to the development of compounds that are about 10 times more active than existing commercial inhibitor blends [2]. Patent applications are being prepared for these compounds.
· Prediction of a new ultra hard phase of titania was predicted from simulations of the bulk phase stability and confirmed in subsequent high pressure X-ray diffraction measurements. This phase is the hardest know oxide material [3].
· Elucidation of the growth mechanism of Zeolite Beta C and Zeolite L [4],[5]. Detailed investigation of the reaction of secondary building units with zeolite crystal surfaces has revealed the importance of pre-forming oligomeric clusters for crystal growth. This work adds to the body of evidence that implicates secondary building unit formation as a key step in crystal growth of microporous materials.
Our
account of these and other applications follows a brief survey of the new
techniques that have been developed over the lifetime of this grant.
Methodological Developments
Our approach has been based on well-established methods of semiclasssical simulations and electronic structure techniques. Code porting and development of novel algorithms for parallel calculations have been a major theme in our work with significant contributions from consortium members at CCLRC Daresbury Lab. (DL) but also strongly supported by the participating university groups. Both development and application work has been further supported by two consortium postdoctoral assistants Drs Plummer (DL) and Sokol (RI) whose main work concerned respectively ii) and iii) of the programmes listed below.
i) Atomistic modelling has proved to be invaluable to our members, as in novel kinetic Monte Carlo (MC) techniques applied to study diffusion at grain boundaries, or in Molecular Dynamics (MD) simulations on passivated and bare gold nanoparticles in supercritical media, with further studies of cluster agglomeration and growth. Key codes including DL_POLY, GULP, MARVINS and DMAREL have been ported and optimised for the T3E platform.
Electronic structure calculations of very complex materials have only become tractable in the last few years, due to the development of massively parallel versions of plane-wave and local-orbital based Density Functional Theory (DFT) codes expressly for massively parallel architectures. Here we describe developments of periodic and aperiodic codes in ii) and iii) respectively.
ii) Rapid advances within
DFT included development and implementation of a new generation of chemically
accurate and numerically stable exchange and correlation functionals, which
have allowed us to undertake extensive studies on the structure and stability
of complex oxide materials, their surface properties and reactivity using plane
wave calculations. Here, we employed the codes CASTEP and VASP, the performance
of which has been optimised, in particular with input from our members and with
support from the whole consortium, with close collaboration with the UK
Car-Parrinello (UKCP) consortium. Furthermore,
the CRYSTAL-2003 code based on Gaussian atomic orbitals was highly optimised on
the Cray-T3E and later ported and optimised on the IBM HPCx system. Our
implementation of hybrid exchange density functional theory had a dramatic
impact on the accuracy of ground state energy surfaces and the description of
low lying excited states [6]
and magnetic coupling [7]
in particular. The efficiency of the parallel code also allowed us to perform
the largest ever calculation using localised Gaussian basis sets for studies of
the protein Crambin. This calculation was of a fully periodic system with
14,000 basis functions while the largest previous calculation even in
non-periodic systems was 4,000 basis functions. This work was performed in
collaboration with groups measuring X-ray diffraction data on the synchrotron
source at Daresbury and was highlighted in the 2002-2003 CCLRC Synchrotron
Radiation Report [8].
Finally, the order-N local orbital DFT code, SIESTA, was ported and
optimised on the T3E and SGI platforms.
The implementation of SIESTA was central to the work described in
section C1 on zeolite surfaces, where the large length-scale and number of
atoms precludes the use of ‘traditional’ DFT methods.
iii) Development of hybrid QM/MM embedding techniques undertaken at the RI in close collaboration with DL (ChemShell) and at UCL (GUESS) has dramatically furthered our capabilities in assessing local, chemical and physical processes at solid surfaces and interfaces. The new codes were successfully ported on CSAR SGI platforms (Fermat, Green and Wren) and employed in very productive studies of surface defects, sorption and catalysis described in section B5.
Applications
A. Crystal Structure, Defects and Transport
A1. The structure elucidation of complex oxide materials (N. de Leeuw; Birkbeck)
We have elucidated the ordering of hydroxy groups in hydroxy-apatite Ca10(PO4)6(OH)2, which is the major constituent of mammalian bones and tooth enamel. Experimentally it was unknown whether the 50% occupancy of the hydroxy sites in the crystal structure was due to true disorder or an averaging of local domains. Our electronic structure calculations showed clearly that there is a considerable energetic incentive for local domain formation rather than disorder of the hydroxy groups over the crystallographic sites [9]. Calculations of solid solutions of fluor- and chlor-apatite, which are important in geological situations, have shown that complete mixing of the two end-members can be expected on energetic grounds, with ordering occurring only in the 50% mixed structure, where columns of F- or Cl- ions in the c-direction are preferred over sheets in the ab-plane [10],[11].
Calculations of the incorporation of water into the scheelite CaWO4 mineral structure have shown that the introduction of water in the lattice leads to 2-dimensional layering of the previously 3-dimensional structure [12],[13],[14]. Although this behaviour shows a superficial similarity to clay-type minerals, it is even more suggestive of the possible existence of hydrous pre-cursor phases to the anhydrous structure, similar to calcium carbonate materials which are formed from hydrous pre-cursor minerals such as ikaite CaCO3.6H2O.
Finally, we have investigated hydrogen-containing defects in SiO2 and MgO and compared these with the same defects in the mixed phase Mg2SiO4 [15],[16]. Our calculations have shown that the mixed material acts like a composite of the two pure oxides and that its structure is similarly a mixture between ionic and covalent, with ionic bonding between the magnesium and oxygen ions, but semi-covalent bonding between the silicon and oxygen ions, which exist in the structure as SiO4 groups.
A2. Computational studies of proton conducting materials (M. S. Islam; Surrey)
In line with the original proposal we have undertaken a programme of computational studies of solid state proton conductors, which are of direct relevance to new perovskite materials for potential use in sensors and fuel cells. The powerful synergy of atomistic simulation and ab initio techniques has provided unique insight into the materials properties.
Here we focused, for the first time, on the distorted orthorhombic phase of CaZrO3, which extends earlier simulation work on ideal cubic perovskites [17],[18],[19],[20]. The ab initio MD calculations using the CASTEP code indicated that proton conduction occurs via a simple transfer of a lone proton from one oxygen ion to the next (Grötthuss mechanism), with no evidence for the migration of hydroxyl ions (“vehicle” mechanism). We also find considerable effect of lattice vibrational dynamics so that for each hopping event the O(H)–O separation shortens so as to facilitate proton transfer. In an attempt to probe the question of proton-dopant association, we have undertaken, for the first time, a series of calculations on dopant-hydroxyl defect pairs in CaZrO3. The resulting energies predict favourable clusters, in accord with proton "trapping" energies derived from recent muon spin relaxation and quasi-elastic neutron scattering experiments. The present calculations therefore predict possible trapping effects, which are analogous to oxygen ion conductivity in fluorite oxides and the well-known importance of dopant-vacancy interactions.
In general, such computational studies form part of the continuing effort to improve our understanding of proton transport, a key phenomenon in a variety of systems that range from inorganic solids to biomolecules.
A3. Coherent control of archetypal reactions in solids (A. Shluger; UCL)
We used theoretical modelling to investigate the feasibility of using two suitably phased and specially tuned ultra-fast laser pulses to break specific bonds, and to drive dissociation reactions in solids to desired products. In particular, we focused on dissociation of an excited crystal bond – self-trapped exciton into a pair of defects in the bulk of an alkali halide crystal. Predicting optimal control parameters for this case turned out to be very challenging as there has been no previous quantum-mechanical modelling of the exciton self-trapping process. To tackle this problem, we first studied the possible rôle of thermal fluctuations of atoms in a crystal as precursor states for exciton and hole polaron self-trapping, and for the initial stages of self-trapping dynamics [21],[22],[23]. Analysing the molecular dynamics of archetypal cubic ionic insulators MgO and NaCl at several temperatures, and calculating their electronic structure for selected atomic configurations, we demonstrated that thermal fluctuations of the electrostatic potential in the crystal ground state create transient localised electronic states. We suggested that these states can serve as precursors for the self-trapping of excitons and can be selectively excited near the optical absorption edge. However, there is no guarantee that excitons trapped initially on these fluctuations will be localised and evolve into self-trapped states.
To model an adiabatic transformation from delocalised to self-trapped exciton state, we applied plane-wave DFT [24],[25]. Previous atomistic modelling has only been successful in calculating the structure and properties of strongly localised systems and it was anticipated that this new technique would provide further insight into this complex problem. Our calculations, however, highlighted severe problems in applications of the DFT to calculations of self-trapped polaron and exciton states in insulators. The local and GGA density functionals yielded solutions biased towards the delocalised states to such an extent that no stable self-trapped state was predicted for either an exciton or a hole. This is contrary to available experimental evidence. We have also demonstrated that the self-trapped solution gradually becomes energetically more stable as the amount of exact exchange is increased in hybrid type density functionals. These results provided a significant insight into the mechanisms and feasibility of coherent control of archetypal reactions in solids using pump-dump schemes and improved our ability to model dynamics of reactions in solids. This approach has recently been extended to studies of electronic and dynamical properties of quantum dots [26].
A4. Calculations on grain boundary diffusion (J.H. Harding; UCL)
Calculations have been performed on grain boundary diffusion for a number of systems. Since the grain boundary diffusion rate is slow, even at high temperatures, it is not practical to obtain it by a direct molecular dynamics simulation. The calculations were therefore performed using a method based on kinetic Monte Carlo using the standard expression for the diffusion coefficient where a number of different trajectories contribute to the process:
![]() ,
,
where Dxx is the diffusion coefficient in the x direction, na is the geometrical probability of a jump of type a, rxa is the projection of that jump on the x axis, Ga is the hopping rate, fxa is the partial correlation function and the summation is taken over all the possible types of jump in the boundary. The individual activation energies are calculated using a drag method to find the transition state as described in [27]. Current work is using the temperature-assisted dynamics methods developed by Voter.
Calculations have been performed for NiO, MgO, Al2O3 and Cr2O3 on simple mirror-twin boundaries. Since the defect concentration depends on the presence of an unknown level of doping, it is impossible to compare our calculations with the experimental pre-exponential factor. We have assumed for convenience a grain boundary width of 5Å. The results clearly show the anisotropy expected from structural considerations and observed in some bicrystals (see ref. [28] for a full discussion). Calculations on the diffusion rates for paths near grain boundaries strongly support the idea of an effective width for diffusion (i.e. the region where the diffusion rate is significantly different from the bulk) of about a lattice parameter, consistent with the value assumed above.
Calculations have also been performed to investigate the effect that isovalent impurities in the grain boundaries of alumina have on the migration energy of defects [29],[30]. There are two effects. The first is a binding energy, which is particularly important for large ions such as La3+. This tends to reduce the concentration of available mobile defects. The second is an effect on the migration energy; it is harder for the defects to move through the boundary in the presence of the dopants. This ‘blocking’ effect is often cited as the explanation of the ‘reactive element’ effect, whereby certain elements are added to alloys to produce thin, dense, protective oxide films.
A.5 Phase Stability and Transport in Solid State Battery Cathodes (N.M. Harrison; DL)
Transition metal oxides are promising electrode materials for advanced high energy density batteries. These materials have relatively open structures and a flexible electronic structure, which facilitates the intercalation of Li-ions. Recent improvements in the efficiency and reliability of first principles calculations, with the CRYSTAL and CASTEP codes, means that it is now possible to predict structure, electronic structure and intercalation energetics for rather realistic models of battery anodes.
Initial
studies of the bulk phase stability of titania lead to the discovery of a
previously unknown, high pressure phase in the cottunite structure which was
predicted to have a very high bulk modulus (~450 GPa). Subsequent preparation
of the phase in collaboration with Dubrovinsky in Upsalla lead to the synthesis
of a new phase which was confirmed to be the hardest known oxide material [3],
[31].
Calculations
of lithiated rutile structures for a wide range of concentrations lead to a new
model of the intercalation kinetics and thermodynamics [32],[33],[34].
Within this new model open circuit voltage profiles were computed for both the
rutile and anatase structures [35]
that were in excellent agreement with those observed and provide a new detailed
explanation of cathode performance. In addition the unusual structural
distortions accompanying intercalation were explained [36].
B. Surface Science and Catalysis
B1. Pd and Pt supported on cubic-ZrO2 and CeO2 (C.R.A. Catlow, M. Alfreddson; RI)
Interfaces where metals and oxides are in direct contact play an important rôle in many industrial applications (e.g. in electronic and optical devices, sensors, solid oxide fuel cells, the exhaust automobile catalyst, composite materials, coatings, and magnetic nanoparticles), stimulating research in the field of metal/oxide interfaces. However, it is not only the industrial applications, but also the desire to control the properties of the metal-oxide bond that drives these studies. In this project we investigated how the metal-oxide bond varies for various substrates (c-ZrO2 and CeO2), surface structures ({111}, {011} and {310} surfaces of c-ZrO2), and different noble metals (Pd and Pt) [37], [38]. We have studied which are the most stable adsorption sites, the mobility of the noble metals on the substrate, and to gain insight into of the catalytic activity of these interfaces we determined the electronic structure of the metal-oxide bond.
We concluded that the most stable adsorption sites for both Pd and Pt on top of c-ZrO2 is the oxygen terminated steps sites or oxygen defects on surface terraces. However, while 1-fold oxygen coordinated sites favour Pt, Pd prefers 2- and 3-folded oxygen coordinated sites. We also find that the Pt-interfaces are thermodynamically more stable than the Pd-interfaces, and that the Pt-Pt bonds are 2 times more stable than the Pd-Pd bond, indicating higher mobility of the Pd than the Pt atoms on the surfaces [37,[39]].
We have also calculated the wetting angles for Pd and Pt on the {111} surface of c-ZrO2, and for 1-monolayer of the noble metal we predict a wetting angle of roughly 110º for both Pd and Pt. Increasing the metal coverage we calculate that the wetting angles also increase to ca. 120º [40].
The adsorption energies of Pd (ca. 200 kJ/mol) and Pt (ca. 400 kJ/mol) on the CeO2 and c-ZrO2 substrates are similar, but we observe different geometrical structures on the two substrates: on c-ZrO2 Pd is adsorbed tilted to the outermost oxygen ions, while on CeO2 Pd is adsorbed directly on top of the outermost oxygen ions. This observation is explained by the more covalent bond of ZrO2 than of CeO2, where the d-orbitals on the Zr ions direct the Pd-O bond [38].
B2. Sorption, Hydration and Surface Defects (N. de Leeuw; Birkbeck: P. Lindan; Kent)
Our wide ranging DFT studies under this heading have included (i) the investigation of water adsorption at low-coordinated surface sites on calcium oxide planes, where the coordination of the surface species was shown to be the major factor determining the mode of adsorption, i.e. dissociative chemisorption or associative physisorption [41]; (ii) the competitive adsorption of water and organic adsorbates at calcium fluoride, which is important for the understanding of mineral separation techniques such as flotation processes [42],[43]; and (iii) the formation of defects at the catalytically important silver (111) surface [44]. These calculations have shown that the formation of adatoms, vacancies and extended steps at the surfaces is not energetically expensive and that the calculated surface energy of the (111) surface is much closer to the experimentally determined value if we take into account these defective surfaces.
Further work employing CASTEP unfolded some of the structural complexities of the oxide/water interface. These calculations have also led to reinterpretation of key experimental results [45],[46]. Related work on adsorption at defect sites [47] and on co-operative effects among co-adsorbates [46] represent considerable advances in meeting the challenge of modelling surfaces and processes realistically.
B3. Nano Structured Carbon (P. Lindan; Kent)
We have pursued a new line of research into nanostructured carbon and its structure-property relationships. We have examined hydrogen adsorption, spin and the role of defects in carbon nanotubes. Our calculations show that chemisorbed hydrogen may induce a magnetic gap state in a nanotube, and moreover this is very sensitive to the presence of ring defects even if they are distant from the adsorption site. Curvature and ring defects also have a pronounced effect on the adsorption energetics, suggesting ways in which the chemical properties of nanostructured carbon may be engineered.
B4. Catalysis on metal and complex oxides (D.J. Willock; Cardiff)
Studies of heterogeneous catalysis require the full electronic structure of solid surfaces and adsorbed reactants to be calculated. We have used the CSAR facility to generate new information on the adsorption and diffusion of hydrogen on the surfaces of the main transition metal hydrogenation catalysts, Ni, Pd and Pt. This work demonstrated that the differences observed experimentally in the response of these catalysts to hydrogen concentration can, in part, be explained by differences in the diffusion of H atoms over the metal surfaces [48]. This work provided simulation input into a collaborative IMI project in which the DFT generated adsorption data [49] were used as input for a micro-kinetic model of alkene hydrogenation.
We have also studied catalysis using oxide materials, in particular the activation of methane over Ga2O3, which forms an important step in the partial oxidation of methane to methanol over mixed Ga2O3/MoO3 catalysts. The yield from this type of catalyst is too low for commercial exploitation but the massive worldwide deposits of methane make it an attractive process for academic study with the aim of improved selectivity avoiding total oxidation. This project was funded by the Japanese RITE foundation and simulation work was carried out in tandem with laboratory reactor studies. Using periodic DFT calculations we have studied the defect structure of the low energy surfaces [50],[51] of this material and demonstrated that C-H bond activation at the defect sites does provide a low energy pathway for generating CH3 moieties. Studying defects using periodic DFT requires large unit cells to be generated to minimise defect-defect interactions and this type of calculation was only possible with the provision of CSAR resources.
In a second IMI funded project we have also used the facility to address the adsorption of ketones to the same set of metal surfaces. The electronegativity of oxygen tends to favour an end-on adsorption mode and we have also addressed the question of keto-enol tautomerism. While the gas phase calculations confirm that the keto-isomer is strongly favoured for ketones such as acetone, our results show that the enolate form becomes the more stable on transition metal surfaces [52]. We have used these results to inform our work on the enantioselective hydrogenation of pyruvates using cinchona modified Pt catalysts. The calculations suggest a different adsorption mode for the pyruvate reactant than had previously been assumed allowing us to suggest a new model for the modifier-reactant interaction [53],[54].
B5. Surface structure and catalysis over ZnO (A. Sokol, S. French, C.R.A. Catlow; RI: A. Wander, N.M. Harrison; DL)
A series of computational studies on ZnO surfaces have been undertaken aimed at elucidation of the bulk and surface properties of this technologically important material with particular emphasis on its applications in catalysis and electronics.
The group at DL concentrated on DFT studies of surface polarity in
ZnO using the CRYSTAL-2003 code, looking at the polar and non-polar surfaces,
at defect structures and at the adsorption properties of catalytically relevant
molecules [55],[56],[57],[58],[59].
Studies of the polar surfaces of MgO and NiO led to an understanding of the
surface stability and its interactions with water [60].
In an alternative and complementary approach (RI group), we made use of DFT techniques (Dmol3 and CASTEP) along with experimental data to parameterise new interatomic potentials, which were then employed in the study of structure and defect properties of both non-polar (10-10) [61] and polar, (0001)-Zn and (000-1)-O terminated, surfaces [62],[63],[64]. Having established the principal surface models, we proceeded to investigate methanol synthesis over polar surfaces of ZnO with hybrid QM/MM embedded cluster techniques previously developed at RI and DL. The full catalytic cycle was studied with the focus on the adsorption modes, atomic structure and energetics of main reactants, stable intermediates and products in the adsorbed state [62,[65]]. To validate our approach, the proposed models and conclusions with regard to the atomistic mechanisms of catalysis, we calculated vibrational spectra of the most stable key intermediates (e.g. hydrogen and formate) and found close agreement with experiment [63,[66]]. Next, we extended these studies to the Cu/ZnO system, which is widely used as the main working component of the ICI methanol catalyst. We started with an investigation into the properties of a Cu atom adsorbed on the (0001)-Zn surface of ZnO and identified vacant Zn surface sites as primary sites for anchoring Cu clusters; next, we concentrated on initial stages of nucleation and growth of the anchored Cu clusters [32,61,[67]]. We are continuing this work using current HPC(x) facilities.
C. Crystal Growth and Nucleation
C1. Modelling zeolite growth (B. Slater; RI)
During the period (2001-2003), we used the CSAR facility extensively to investigate fundamental aspects of zeolite growth, employing both classical and electronic structure techniques. In our initial work we focused on the intersecting 12MR material, zeolite beta C; in this work we used classical modelling techniques to deduce the most stable terminations for the {100} surface and then considered the reaction of a number of secondary building units with the zeolite surface. By evaluating a number of reaction pathways, we deduced that single 4 ring and double 4 rings play an important rôle in dictating the growth of this material [3,[68],[69]]. More recently, we applied similar methodology to examine the growth of zeolite L, an important material with catalytic and nano-container applications. We found that careful examination of the possible assembly mechanisms suggests that the crystal habit, which varies from thin hexagonal plates to thick rods, is controlled by double 6 ring and cancrinite cage fusing kinetics at distinct crystal faces [4,[70]]. The results of the zeolite L study are particularly relevant to membrane applications, where manipulating the aspect ratio, which changes the surface area of channel systems expressed on the crystal surface, is a vital part of crystal engineering. Recently we have built upon the zeolite beta C work to examine metastable stages of growth, for a given surface using newly developed software [70,[71]]. This work opens up new insights in the processes involved in growth, and the delicate balance between thermodynamic and kinetic factors during crystal assembly.
C2. Computational insights into agglomeration between crystal surfaces in solution (S. Price; UCL)
This grant provided the computational resources for some of the larger Molecular Dynamics studies required for a collaborative investigation of the factors that influence agglomeration of crystallites in industrial crystallisation process, funded by EPSRC as part of the Chemists and Chemical Engineers programme. Profs. Jones and Simons and F. Pratola of UCL Chemical Engineering, measured the forces required to break the agglomerative bonds formed between various potash alum crystal surfaces. We provided atomistic models for these surfaces, by determining the stable structures of potash alum in saturated solution. Since the [111] face of this complex hydrated crystal is polar, this required innovative modelling of dipolar, charged and reconstructed surfaces corresponding to various cuts through the crystal.
In order to comment on the forces involved as crystallites approach each other in solution, prior to the formation of the agglomerative bond, simulations were performed on two nano-crystallites of potassium chloride in solution. The forces between the crystallites over the last nano-metre of separation vary from attractive to repulsive, with a strong dependence on the saturation of the solution, and the speed of approach and ability to move laterally. The atomistic picture of the hydrated ions affecting the forces and their ability to diffuse from the slit pore depending on the relative motions of the crystallites has provided insights into the difficulties inherent in predicting agglomeration propensities, even when the structure of the surfaces is known.
C3. MD simulation of metal nanoparticles in supercritical media (M.Lal; Liverpool, M.Plummer, W.Smith; DL)
Under this project, we have pursued MD simulations of systems of bare and passivated metal (in particular, gold) nanoparticles in supercritical solvents to gain molecular level understanding of the efficacy of such media in the size-selective precipitation, a most promising process for isolating particles of a given size from polydisperse samples. Since solvent quality plays a vital rôle in the process of size-selective precipitation, our simulations have been directed to investigating the effects of the particle size and the presence of the passivating layer on the mode and the degree of solvation of the particles in the vicinity of the critical point of the solvent.
Simulations of 8-atom and 38-atom, bare and passivated, gold nanoparticles have been performed in ethane along the critical isochore in the vicinity of Tc. The passivating ligand featuring in the model is dodecane thioate, attached to the particle surface by the Au ─ S bond. The 8-atom and the 38-atom passivated particles had, respectively, 8 and 24 ligands bonded to their surfaces. The simulation results are consistent with the experimental observations that in the phase region considered the solvent quality, represented by the degree of solvation, deteriorates with the increase in the particle size. The presence of the passivating layer reduces the degree of solvation. For the 8-atom passivated particle, the solvent quality passes through a minimum as the system moves from the sub-critical to the supercritical regime. The configurational structure of the passivating layer plays a crucial rôle in determining the temperature variation of the degree of solvation and hence of the solvent quality [72].
C4. Clathrate Hydrates (P.M. Rodger; Warwick)
Simulations with the hardware provided by this grant have enabled at least three major developments in the modelling and understanding of clathrate hydrates. In the first place, computational screening methods have enabled us to identify a completely new class of low dosage inhibitor for clathrate hydrate formation [1]; subsequent development of the new lead compound has lead to the development of compounds that are about 10 times more active than existing commercial inhibitor blends [2]. Patent applications are being prepared for these compounds.
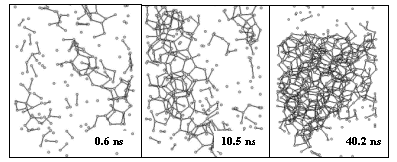
Figure 1. Growth of methane hydrate crystal in molecular dynamics simulations
Long timescale simulations using the cluster have enabled us to produce both the first simulation of primary nucleation of methane hydrate crystals (see Figure 1) [73], and the first direct simulation of low dosage inhibition [74]. The latter was achieved using polyvinylpyrrolidone (PVP) with molecular weight comparable to those used in industrial applications (ca. 1000 u); PVP is often taken as the archetype for low dosage hydrate inhibitors. The power of the national facilities has also enabled us to undertake the first detailed study of polydispersity of the inhibitor, with a range of molecular weights and tacticities being simulated to search for any correlation with inhibiting power.
These simulations have been instrumental in revising our understanding of the mechanism for nucleation. In particular, they have identified shortcomings in the current understanding of the molecular mechanism for both nucleation and low dosage inhibition. Most strikingly, they have shown that a strict understanding of surface adsorption is not essential for low-dosage anti-freeze activity in hydrate inhibitors. These simulations have enabled us to develop a new mechanistic understanding of both the formation and inhibition processes that is consistent with both recent experiments and with the new simulation data [75].
The publications arising from this grant, and the references, can be found at http://www.ri.ac.uk/mcc.
Summary
Large-scale simulations using MPP resources has enabled the study of a wide range of fundamental topics in materials science including transport properties, catalysis, reactivity, nucleation and crystal growth, with a growing emphasis on understanding non-equilibrium processes. Our work has been exceptionally productive leading to more than 80 publications to date in leading journals and attracted further international recognition. The code optimisation work undertaken during this grant has resulted in the development of transferable expertise in optimisation/algorithms that has benefited a number of MPP consortia and the materials simulation community in general. In addition, because of the foundation work on the CSAR resource, we have found that developments instigated during the CSAR project have accelerated our rate of scientific progress on the HPCx facility. The scientific achievements described here coupled with lasting and ongoing contributions to atomistic materials science code development indicate the excellent value for money this grant has provided.