We don't do much on molecular evolution in this course. This is in part because so many other courses deal with molecular genetics, and indirectly with its evolution. It is also partly because we feel that understanding how evolution works is as important than the material (DNA, protein, phenotype) on which the evolution is based. And you probably get many other courses that have molecular information. We aim to provide some of the basis for you to go on and understand evolution in any system, including in molecules.
But there is no doubt that the biggest
sector of evolutionary biology around today is molecular. So I want to
spend this lecture trying to link this course into the curriculum of every
modern geneticist. Also, it gives me a chance to introduce you to the extraordinary
kerfuffles created in evolutionary biology by discoveries in molecular
genetics!
The "Classical School". Before the late 1960s, it was generally believed that polymorphism (the presence of more than one allele in a population of genes) was relatively rare, and that evolution consisted mainly of:
1) mutation (mostly deleterious)The "Balance School". A few people, such as "Dobzhansky, who began his career collecting beetles in Central Asia, and the British School ... who carried on the genteel upper-middle-class tradition of fascination with snails and butterflies" (Lewontin 1974: 30), believed that polymorphisms were fairly common, and indeed important. They believed and indeed showed that morphological and chromosomal polymorphisms, for instance, were retained because of heterozygous advantage or other forms of "balancing" selection.
2) if advantageous, increase to fixation (or loss due to deleteriousness).
The molecular biology revolution. In the mid-1960s, Lewontin and Hubby in the USA working on Drosophila and Harris and Hopkinson here at UCL (Wolfson House) working on humans discovered that electrophoresis could reveal enzyme polymorphism using staining techniques developed from microscopy. To everyone's surprise, many enzymes had enormous amounts of polymorphism, far more than expected by the classical school. However, the balance school felt that all the polymorphism could be explained as due to selectively maintained genetic variation.
Genetic load and problems for the "Balance School". However, Lewontin and others argued that to maintain such a high amount of genetic diversity would be too costly. If heterozygotes at thousands of genes in the genome were advantageous, this would result in the production of a lot of homozygotes in every generation. A lot of (1-s) and (1-t) values of fitness for homozygotes must combine in a single individual, to produce an extremely heavy "genetic load". For example if a mere 1000 genes were homozygous (and there are probably 30,000 - 50,000 in Homo sapiens), each with a selection coefficient of 0.01, the fitnesses per locus would be 0.99, and (0.99)1000 = 0.00004 over the whole genome.
Even supposing outbreeding populations managed to survive such enormous genetic loads, imagine what inbreeding would do! Inbreeding decreases the homozygosity by a fraction F. In sib-sib matings, F = 0.25. Supposing 10,000 genes are polymorphic, each with heterozygosity (i.e. 2pq) of 10%. Then within each individual, there will be on average extra homozygosity of 2Fpq, or 10% x 0.25 = 2.5%. This means about 250 extra genes are homozygous out of the 10,000. If each suffers a 1% disadvantage, the total fitness of an inbred individual will be 0.99250, equivalent to 8% survival of inbred offspring. Now sib-sib matings are prone to inbreeding problems, but a 92% greater mortality rate for inbred offspring seems too high.
There are many difficulties with these genetic load arguments, but I won't bore you with them. However, suffice it to say that even weak selection maintaining large numbers of polymorphisms doesn't seem very friendly to the homozygotes that must result in every generation. Think of sickle-cell anaemia and thalassaemias! If most polymorphic genes were like that we would be in a lot of trouble!
The neutral theory and molecular clock: the "Neo-Classical School". Two papers, by Motoo Kimura, and by Jack King & Thomas Jukes, are generally recognized as the key promoters of the "Neutral Theory", the "new wave" in molecular evolution. By the late 1960s, amino-acid sequences for proteins from a number of taxa were known, and the full implications for evolution of the degenerate DNA code for amino acids became apparent.
Kimura (1968) made calculations based on
knowledge of amino acid sequences in ![]() -
and
-
and ![]() -haemoglobin
in man and other mammals. To explain the observed rates of amino acid substitutions
(about 1 a.a. substitution for each 100 amino acid polypeptide per 107
years), we require in mammals over the whole genome an overall nucleotide
substitution rate of about one nucleotide substitution per 2 years. Kimura
argued that DNA evolution at this rate couldn't possibly be explained by
selection. Haldane had made an earlier genetic load argument to show that
new alleles could not be substituted via selection much faster than one
every 300 generations (about 1 every 1200 years in humans!) without driving
the population extinct due to genetic load. Kimura therefore argued that
most of this variation and evolution must be neutral, and that slow, random
genetic drift at very large numbers of simultaneous DNA polymorphisms was
responsible. King and Jukes (1969) in a paper entitled "Non-Darwinian Evolution",
amplified Kimura's hypothesis.
-haemoglobin
in man and other mammals. To explain the observed rates of amino acid substitutions
(about 1 a.a. substitution for each 100 amino acid polypeptide per 107
years), we require in mammals over the whole genome an overall nucleotide
substitution rate of about one nucleotide substitution per 2 years. Kimura
argued that DNA evolution at this rate couldn't possibly be explained by
selection. Haldane had made an earlier genetic load argument to show that
new alleles could not be substituted via selection much faster than one
every 300 generations (about 1 every 1200 years in humans!) without driving
the population extinct due to genetic load. Kimura therefore argued that
most of this variation and evolution must be neutral, and that slow, random
genetic drift at very large numbers of simultaneous DNA polymorphisms was
responsible. King and Jukes (1969) in a paper entitled "Non-Darwinian Evolution",
amplified Kimura's hypothesis.
Supposing the neutral mutation rate for
a given protein was ![]() , and the
number of alleles in a diploid population is 2Ne (Ne
is the effective size of the population), then the number of new mutations
in every generation is 2Ne
, and the
number of alleles in a diploid population is 2Ne (Ne
is the effective size of the population), then the number of new mutations
in every generation is 2Ne ![]() .
These neutral alleles will either drift into the population, or (mostly)
will simply be lost almost immediately. Given the 2Ne
neutral alleles in the population, what is the probability that each one
will eventually be fixed as a substitution? One of the alleles will eventually
be fixed, but since each has identical fitness, each has the same probability,
1/2Ne. Thus the rate of origin of neutral alleles that
will eventually be fixed (which must also be the approximate rate of fixation)
in the population is 2Ne
.
These neutral alleles will either drift into the population, or (mostly)
will simply be lost almost immediately. Given the 2Ne
neutral alleles in the population, what is the probability that each one
will eventually be fixed as a substitution? One of the alleles will eventually
be fixed, but since each has identical fitness, each has the same probability,
1/2Ne. Thus the rate of origin of neutral alleles that
will eventually be fixed (which must also be the approximate rate of fixation)
in the population is 2Ne ![]() /2Ne
=
/2Ne
= ![]() . The rate of origin and the
rate of eventual fixation must be approximately the same, so the rate of
fixation of neutral mutants will also be
. The rate of origin and the
rate of eventual fixation must be approximately the same, so the rate of
fixation of neutral mutants will also be ![]() .
.
This startlingly simple new theory suggested that the rate of neutral molecular evolution was independent of the population size, and was simply equal to the neutral mutation rate.
The time to fixation (from origin of the
mutation that will eventually be fixed until its ultimate fixation under
pure drift), will of course be long: "diffusion theory" shows that this
fixation time averages simply to 4Ne generations. Whereas
the rate of origin and fixation (=![]() )
of new mutations is independent of population size, the rate of progress
through the population is therefore directly proportional to population
size. In a large population, it takes a very long time. For example, in
a population of size 1 million, it takes 4 million generations. Under the
neutral theory, then, the polymorphisms in large populations are due to
a lot of neutral changes originating, and ultimately being lost, but passing
through at such a slow rate that there are usually several alleles at any
particular protein drifting through the population at the same time.
)
of new mutations is independent of population size, the rate of progress
through the population is therefore directly proportional to population
size. In a large population, it takes a very long time. For example, in
a population of size 1 million, it takes 4 million generations. Under the
neutral theory, then, the polymorphisms in large populations are due to
a lot of neutral changes originating, and ultimately being lost, but passing
through at such a slow rate that there are usually several alleles at any
particular protein drifting through the population at the same time.
"Neutralists" like Kimura and King and Jukes, therefore, didn't deny that Darwinian evolution via natural selection occurred. They simply thought that these advantageous mutations, or "balanced polymorphisms" were extremely rare. Most polymorphisms, they argued, must be "effectively neutral". The probability of fixation of a neutral allele by drift is 1/2Ne, and if this is bigger than typical selection pressures, drift will outweigh selection. Thus the neutral theory doesn't even argue that most molecular polymorphisms are completely neutral, just that any selection pressures may usually be outweighed by drift.
If the neutral theory is correct, we might expect to see more rapid evolution in regions of the genome not constrained by selection than in regions strongly constrained by selection. This is in general true: synonymous changes occur much more rapidly than non-synonymous changes overall. Thus most of the evolution seems to be due to base pairs that are not connected strongly with the phenotype, and most likely to have little effect on fitness.
The "molecular clock" argument, was first
proposed by Zuckerandl & Pauling in 1965. Empirical data on protein
sequences showed that amino acid evolution (i.e. substitution) was linear
with time. Neutral theory could explain this, of course: if ![]() was approximately constant across species, and if most protein evolution
were neutral, then the rate of evolution should be roughly the same in
all lineages.
was approximately constant across species, and if most protein evolution
were neutral, then the rate of evolution should be roughly the same in
all lineages.
Today.... These simple arguments are now known to be too simple. The molecular clock doesn't tick at a perfectly constant rate, in part because there are variations between genes, between different parts of lineages, and in part because with different generation times, mutation rates, and possibly even a bit of natural selection, it is all messier then was thought at first. As we shall see, there seems to be a lot more selection on molecular polymorphisms than we once thought.
However, the neutral theory and the molecular
clock arguments are still enormously useful because they are so
simple. Somewhat like the Hardy-Weinberg, these ideas still form the "null
hypothesis", or fundamental basis of all further molecular evolution studies:
the neutralists have had an important and very useful effect on modern
evolutionary biology. The molecular clock isn't perfect, but it still gives
rough estimates of time since divergence, given some sort of calibration
of phylogenies with geological time.
With the advent of DNA technology, we didn't just discover the nature of the genetic material and the genetic machinery which made proteins possible. An entirely new can of worms was opened with lots of new evolutionary problems of its own. Most of these modes of mutation and evolution are familiar to you 2000 + undergraduates, so I will only briefly run through some types of mutational/evolutionary change. It is hard to remember now that there was no inkling that these evolutionary modes even existed before molecular techniques became established.
Molecular evolution and/or mutational modes
Single base pair changes, substitution
Non-coding regions: introns, 5' & 3' flanking regionsPseudogenes
Synonymous
Non-synonymous, amino acid replacements
Genomic evolution
Codon usage biasUses of DNA Technology and molecular evolution
Exon shuffling
Gene duplication, multi-gene families
Whole genome duplication
Phylogeny reconstruction - understanding
the family tree of life.
Conservation biology - saving species
may require knowing who they are related to, how they evolved, and whether
they are inbred
Forensic science - identification
of suspects requires population genetics knowledge; for instance, particular
markers may have different frequencies in different ethnic groups. Positive
identification of suspects must depend on knowledge of the frequencies
in each group.
Behaviour studies - understanding
relatedness in natural populations is now much more firmly based on genetic
markers than hitherto. Genetic analysis of offspring show that females
of many birds that breed in pairs shows that they often have multiple partners;
they indulge in "extra-pair copulation". This is also true for human families:
I believe that a typical "non-paternity rate" in humans is around 20%.
Variable rates in different proteins; severely
conserved in some such as histones, high rates in others, such as some
structural proteins like albumin.
Differences between synonymous and non-synonymous
rates
dS and dN.
Differences between coding regions and
non-coding regions.
Molecular evidence of selection for polymorphism. Alcohol dehydrogenase (Adh) in Drosophila melanogaster. One of the earliest recognized enzyme polymorphisms. Two main alleles, "fast" and "slow", recognized by the speed of migration on the gel. Studies had suggested that the Adh polymorphism were associated with particular environments, and that heterozygous might be important, but these experimental studies were hard to evaluate because Adh was of course embedded in the genome, and linked loci may have been responsible.
In 1983, the first study of DNA sequence
polymorphisms was done on Adh by Martin Kreitman in Lewontin's lab.
Kreitman had sequenced 11 copies of the 2721 bp Adh region, 6 copies
of slow and 5 of fast. Kreitman found 47 DNA polymorphisms:
| no. of variable sites | type | out of | region |
| 18 | single nucleotide polymorphism | 755 bp | introns |
| 8 | single nucleotide polymorphism | 1102 bp | other untranslated |
| 14 | synonymous single nuc. polymorphisms | 765 bp | exons |
| 6 | indels (insertions or deletions) | all in non-coding regions | |
| 1 ONLY!! | NON-SYNONYMOUS single nuc. polymorphisms | (position 1490, fast/slow site) |
In only one case, at position 1490, was there a polymorphism that affected protein sequence, and this was the fast/slow site itself!
However it wasn't until 8 years later that
firm molecular evidence was produced to implicate selection at this protein
polymorphism (Kreitman & Hudson 1991). A comparison of substitution
differences at Adh between the species D. melanogaster and
D.
simulans gave evidence for some variation in the rate of evolution
(presumably due to variability in ![]() )
along the region. Then the expected level of polymorphism could be calculated
(per sliding 100 bp windows). The observed level of polymorphism per 100
bp does not greatly differ from expected except for a few hundred base
pairs around the fast/slow site. The elevated DNA polymorphism in
this region is most likely due to linkage disequilibrium of the synonymous
sites with the
fast/slow site maintained as a polymorphism, so that
these non-coding changes cannot drift to fixation except on their own allele
(fast or slow). This seems good evidence for selective maintenance
of the polymorphism.
)
along the region. Then the expected level of polymorphism could be calculated
(per sliding 100 bp windows). The observed level of polymorphism per 100
bp does not greatly differ from expected except for a few hundred base
pairs around the fast/slow site. The elevated DNA polymorphism in
this region is most likely due to linkage disequilibrium of the synonymous
sites with the
fast/slow site maintained as a polymorphism, so that
these non-coding changes cannot drift to fixation except on their own allele
(fast or slow). This seems good evidence for selective maintenance
of the polymorphism.
There is a variety of other similar evidence for selective maintenance of polymorphisms. A good example is the maintenance in humans of MHC polymorphisms. Many of the MHC polymorphisms seem to have been maintained through the speciation event between humans and chimps, suggesting they were maintained in both ancestors and descendants as polymorphisms. At many other loci, different polymorphisms have been substituted in lineages leading to the humans and chimps We have already met the MHC complex in "Evolution at more than one gene"; the loci in this complex play an important role in disease resistance and are probably under diversifying frequency-dependent selection as a result. Thus observed "transspecific" polymorphisms are likely to have resulted (Futuyma 1998 p. 632).
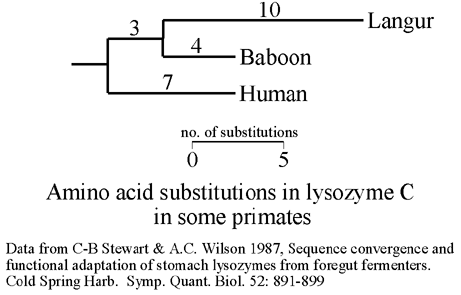
Detection of "positive
selection" via ![]() =
dN/dS > 1
in phylogenies. A number of proteins are now known to be under
"positive selection" (i.e. selection for rapid changes in protein sequence):
lysozyme C is normally a defensive enzyme against bacteria, but in cows
and other ruminants, the enzyme has been coopted for use as a digestive
enzyme in the stomach. In ruminants, lysozyme C is used in digesting bacterial
cell walls, needed for cellulose digestion in the stomach. In the lineage
leading to the langur monkey, which feeds on leaves and uses ruminant-style
fermentation to digest cellulose, there has been rapid evolution of the
lysozyme protein. There is also convergence in amino acid sequence to cow
lysozyme C.
=
dN/dS > 1
in phylogenies. A number of proteins are now known to be under
"positive selection" (i.e. selection for rapid changes in protein sequence):
lysozyme C is normally a defensive enzyme against bacteria, but in cows
and other ruminants, the enzyme has been coopted for use as a digestive
enzyme in the stomach. In ruminants, lysozyme C is used in digesting bacterial
cell walls, needed for cellulose digestion in the stomach. In the lineage
leading to the langur monkey, which feeds on leaves and uses ruminant-style
fermentation to digest cellulose, there has been rapid evolution of the
lysozyme protein. There is also convergence in amino acid sequence to cow
lysozyme C.
Recently, detection of "positive selection" has become much more sophisticated. Here is the general procedure:
1) Sequence copies of the gene of interest from a variety of related speciesHowever, these methods have many pitfalls. DNA has only 4 potential base-pairs at each site, and because some changes are much more likely than others (for example transitions are usually commoner than transversions) substitutions are prone to "homoplasy" (i.e. the same changes occurring in the same sites in multiple lineages), or to multiple changes occurring at each site. So in general we can't just compare DNA sequences in two species, we have to correct for multiple hits. Another problem is that only some parts of the molecule may be under strong diversifying selection leading to
2) Construct a phylogeny (based on genealogy of the gene concerned, or other data)
3) Identify non-synonymous and synonymous changes between the sequences. Estimate dN and dS, and their ratio= dN/dS.
4) Obtain gene-wide estimates of
5) If
Recently, there have been a number of improvements, many of them due to Z. Yang here at UCL:
1) Make more realistic models of molecular evolutionSee example below.
2) Test these models with the data to get better estimates.
3) Estimate multiple
4) Finally, estimate the "posterior probabilities" (a measure of the likelihood) of different values of
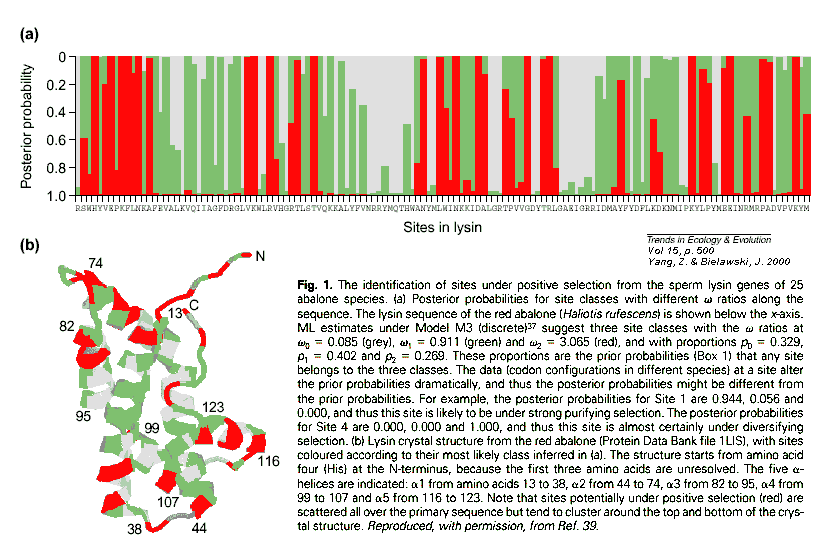
However, "positive selection" (i.e. diversifying
selection) isn't the only kind of selection, and these methods still require
large numbers of replicate sites in order for selection to be detected:
the method it cannot easily detect selection at a few sites embedded in
a strongly conserved region. Probably there is much more selection on molecules
than these estimates of ![]() indicate.
indicate.
The situation at present is that selection seems to be important and detectable in many more proteins than were perhaps expected by the neutralists.
What about genetic load? We still do not have a good idea exactly how many proteins are under this kind of selection, or what the strength of selection must have been: many of the molecules that have been examined were looked at carefully because it seemed likely that they would be under strong selection, an unfair sample, perhaps!
In conclusion, the extent of selection
on proteins, and associated theoretical problems associated with still
needs to be worked out!
Molecular techniques, starting with protein electrophoresis, more recently DNA methods, came online beginning in the 1960s.
Researchers made a major discovery. There was far more polymorphism than expected at the level of genes.
Classical school evolutionists believed that most evolution by natural selection should have led to fixation of mutations that were advantageous; low levels of polymorphism should have resulted.
Attempts to explain this superabundant polymorphism led to the neutral theory and ideas that neutral evolution might provide a molecular clock.
Natural selection: the demise of the neutral
theory
Balancing selection
Positive selection
But neutral theory still provides an important
baseline against which to judge models of selection.
We now know there are many types of molecular evolution.
Theory and data relevant to molecular evolution has many uses.
Next year. If you are interested in a whole course in molecular evolution, you could take Ziheng Yang's course (BIOLC337 Molecular Evolution).
Further reading:
General
textbooks: (Futuyma, Freeman & Herron etc.)
Kimura,M (1968): Evolutionary rate at
the molecular level. Nature 217, 624-626.
King,LC; Jukes,TH (1969): Non-Darwinian
Evolution. Science 164, 788-798.
R.C. Lewontin (1974): The Genetic Basis
of Evolutionary Change. Columbia University Press.
Kreitman,M; Hudson,RR (1991): Inferring
the evolutionary histories of the Adh and Adh-dup loci in
Drosophila
melanogaster from patterns of polymorphism and divergence. Genetics
127, 565-582.