Validation and Deposition
Validation is the process of checking that your model is consistent with stereochemical standards. Most commonly bond angles and bond lengths are defined in a paper by Engh & Huber, 1991 Acta Cryst A47:392-400. Beyond this the source for such information is actually a bit scarce, however there are plenty of reviews that try and draw conclusions based on high resolution structures (e.g. see McArthur & Thornton (1996) Deviations from Planarity of the Peptide Bond in Peptides and Proteins, J.Mol.Biol. 264:1180-1195). Alternatively you can look in the dictionaries currently in use by CCP4 or CNS for the values that the refinement programs use.
If you are model building you can check how closely your model matches general stereochemical principles by looking at the Ramachandran plot (most graphics programs have this function). Additionally coot allows you to check all sorts of other parameters using options in its "Validate" menu:
Although graphics programs do contain validation functions, I find it best to use the software on the pdb deposition website as this represents the criteria you will have to meet before a structure can be accepted into the international database. The following instructions are for the American RCSB portal which I find the easiest to use. It can be accessed by going to the "ADIT" validation server (under the "Deposit and Validate" menu item on the left of the RCSB homepage).
ADIT
In a web browser navigate to the page: http://deposit.pdb.org/validate/
Select "X-ray" in the drop-down menu on the first screen and then click BEGIN to get to the following page:

You can select a pdb file by clicking on the Browse... box next to "Enter coordinate file name" and navigating to the pdb file you want to validate. You need to make sure you select PDB and then Validate next to "Choose Operation". Do not worry about the Precheck option as this always complains about your file but does not seem to influence the validation process.
You can also choose a validation procedure that checks your electron density and structure factors by entering a compressed structure factor file in the second box. However, as it is not yet compulsory to deposit your experimental data I do not worry about this.
Once you have made the appropriate selections and clicked BEGIN (and assuming it has found your pdb file OK), the website will run your data through a program called PROCHECK (and SFCHECK if you gave it structure factors) before presenting you with a results page:


This report is fairly self-explanatory and highlights close contacts, bond distances & angles, torsion angles, chirality, solvent, missing atoms and extra atoms. Generally the best thing to do is open your molecule in coot and then check out each of the atoms/residues flagged up in this report. If you have a very high resolution structure there will probably not be many things flagged in this report, however generally the lower the resolution, the more problems you are likely to encounter. You need to solve all the issues flagged here or else your finally deposited pdb file will report problems in its header for everyone to see.
Sometimes fixing problems and then running the data through a refinement program will leave you with the same issues second time round. Thus if you are sure you are working on your very last version of your model, make the changes in a graphics program, and then re-run the new file through validation without running a refinement program.
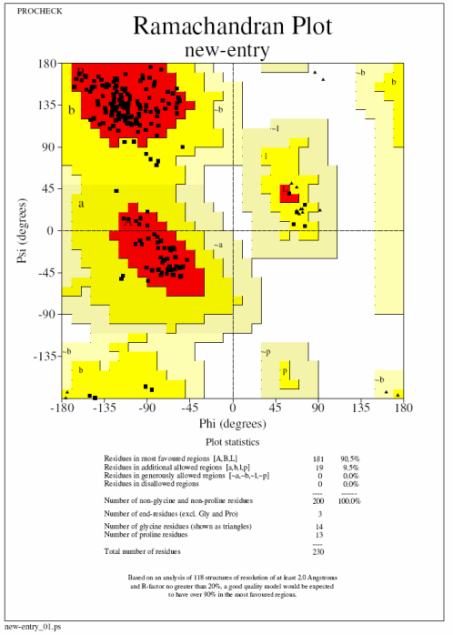
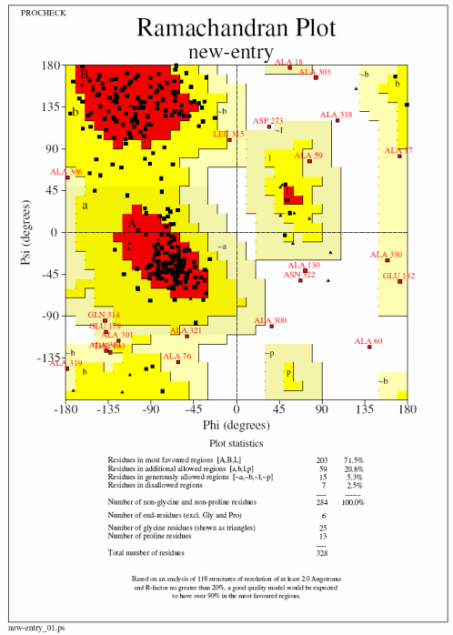
In these plots a square represents a normal amino acid, a triangle a glycine residue, red the most favoured region and yellow additionally allowed regions. Ideally you want to see 90% of your residues in the most favoured regions and none in the disallowed (white) regions (procheck labels any residue in this region so you know which ones to go back and have a look at). The plot on the left (top) is what you are aiming for with no residues in the disallowed and ideally very few in the generously allowed regions. The plot on the right (bottom) is from a 3A structure and has 22 residues in disallowed regions. If you get a bad plot like this it is probably best to run the structure through a refinement program with very tight geometric restraints (ie 0.01 in Refmac).
N.B. The ADIT validation server does not store your data so you can run it as many times as you want!
Running the non-web version of PROCHECK
PROCHECK is also installed on our computer network and can be run by typing procheck followed by the pdb file name and the resolution. Furthermore if you have the file procheck.prm in the directory where you would like to run the program (download it from here and save as procheck.prm), you can change various options. I would definitely recommend scrolling to the end of the file and selecting "Y" to the option "Add 9-character descriptions..." before you run procheck in this way as otherwise it can be hard to tell what is in the files that it outputs. Once you have done this run the program by typing:
setccp4
procheck myfile.pdb 1.8 (obviously you put in your pdb file and resolution here)
To view the postscript output files use xpsview on the sgi's (xpsview file.ps) or KGhostView (in the graphics menu) on the Sun's.
Deposition
Our group policy is only to deposit structures relating to published papers. Similarly we do a lot of research that is subject to strict IP meaning that your structure is not intended to be available to the public. Thus DO NOT DEPOSIT YOUR STRUCTURE unless you have specific permission from Steve or Jon.