Phaser
Phaser is probably the newest and best molecular replacement program around at the moment. It uses maximum likelihood methods for its translation and rotation function searches. For more detailed information about the program, how it works, and a FAQ see the Phaser website.
Although available as a stand-alone program, it can be run from the CCP4i GUI:
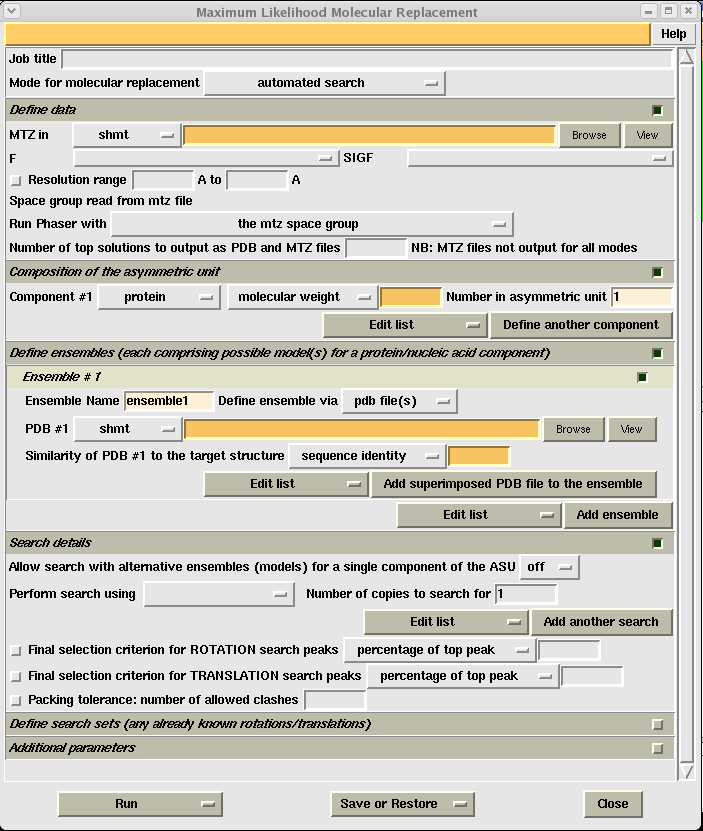
Open CCP4i, choose the "Molecular Replacement" menu on the left and click on the "Phaser" button. This will give you the Phaser input window:
As with other CCP4i programs you need to give each job a title and then fill in all the yellow boxes. You will want to use the MTZ file that came out of the scaling step. Again you need to know the molecular weight of your protein, and the contents of your asymmetric unit (calculated during scaling).
Choosing a search model
Molecular replacement works by rotating and translating a set of protein coordinates within your experimental unit cell that are similar in structure to the protein you are trying to solve. In order to get this to work it is vital that an appropriate model is chosen. In my experience experimenting with different models is almost always the best way to find a successful molecular replacement solution. The following is a list of models I would work through when trying to solve a structure. In all cases I would first edit the search model's pdb file (using a text editor such as nedit or Crimson editor) and remove all waters, metals, sugars and ligands so that just protein atoms are being used.
Running Phaser
Once you have chosen your search model you need to select it in the orange PDB #1 box, and then give an estimated sequence identity as a percentage in the second orange box. IMPORTANT - after you have done this, you need to select "Perform search using ensemble1" (or whichever ensemble you want to search for) under the heading "Search details". You also need to select the number of copies to search for in the asymmetric unit. If you are using suggestion 1 above, this would just be one. However, if your search model is just a single monomer and you expect more than one in your asymmetric unit, you need to tell phaser this explicitly.
Once all this information is filled in you simply click "Run" and then "Run now" (or "Run&View Com File" if you want to see the commands the program is using). Phaser can take anything from twenty minutes to a couple days depending on how big your search model, unit cell and asymmetric unit is.
Assessing the Results
The following diagram from the Phaser manual illustrates the different things Phaser does during an automatic run:
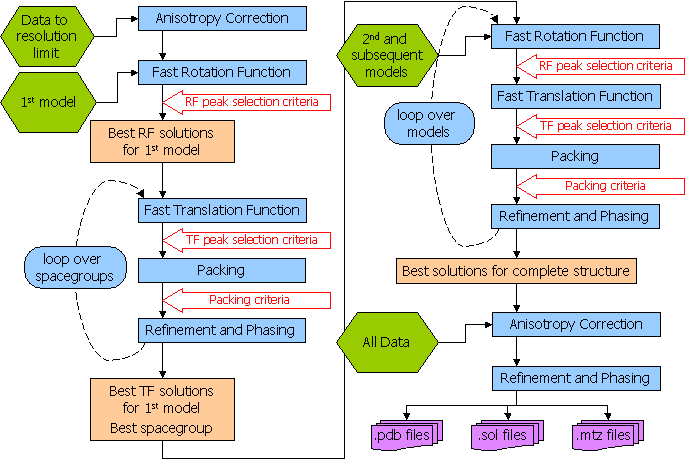
You can specify certain bits of the procedure from the drop-down menu at the top of the Phaser input box, however I have never found much benefit in doing this.
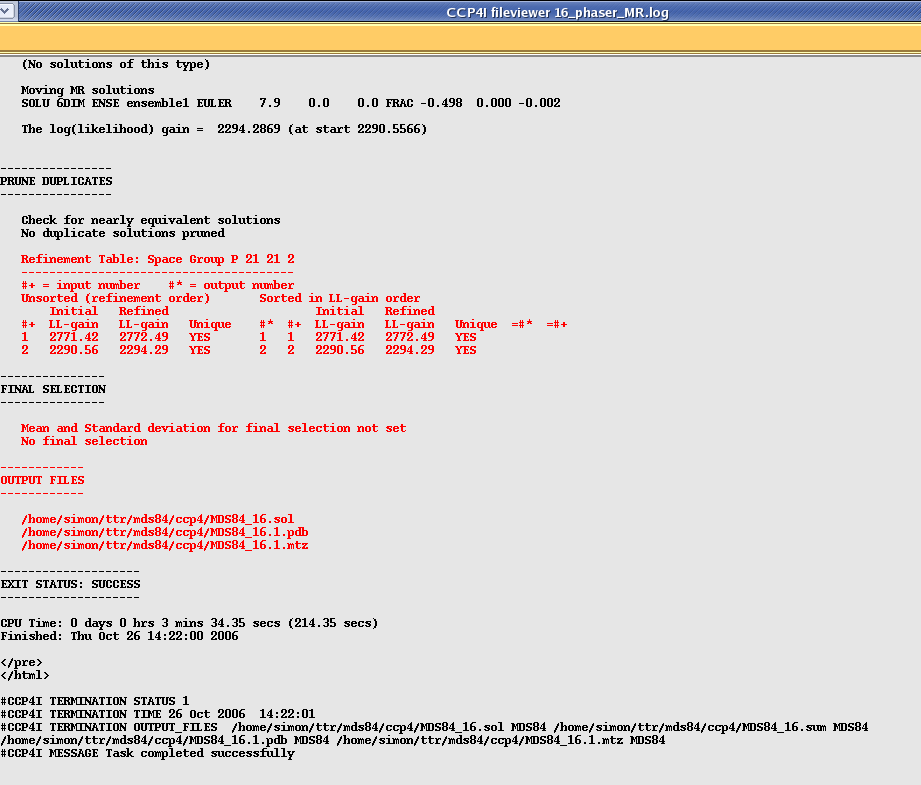
Once your run has finished select the Phaser job and then click on "View Files from Job" and "View Log File". This will show you quite a lengthy file, however each section is clearly labeled as per the scheme represented in the picture above. Scroll down this file until you get to the section that says "Phaser Module: CELL CONTENT ANALYSIS BY MATTHEWS COEFFICIENT", which should look something like this:
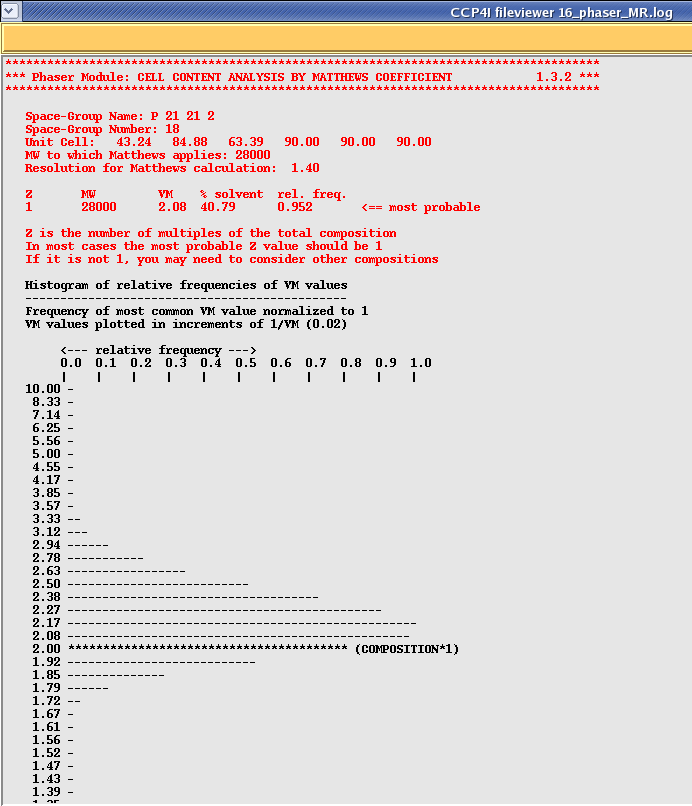
Phaser re-calculates your solvent content for you, and then tells you whether your prescribed search is looking for enough molecules in the asymmetric unit. Note that the histogram is only relative to previously solved structures, however you might want to start asking questions about your model if it is a long way from the mean.
Next scroll done until you get to the results of the rotation function search. This should look something like this:
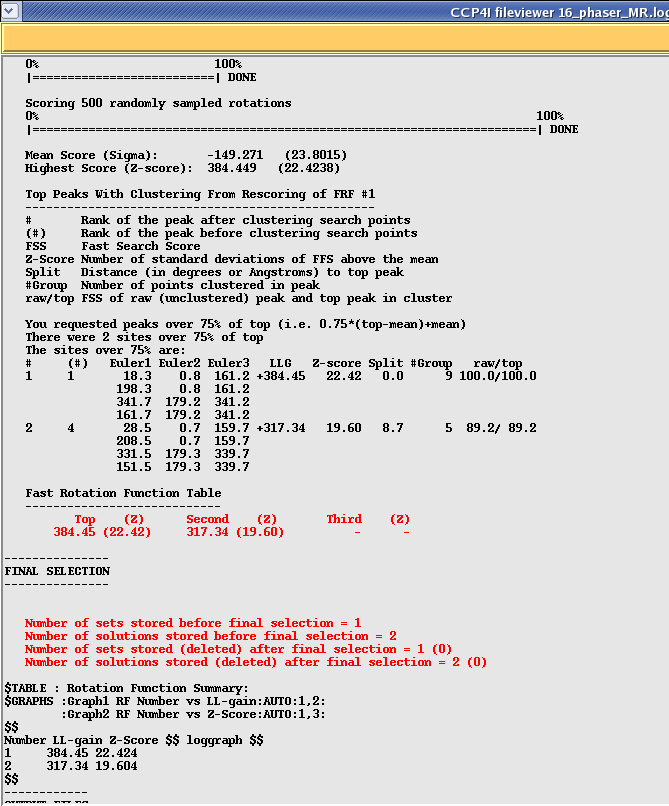
In this example Phaser has found two Rotation solutions. You can tell how strong each solution is by looking at the Log Likelihood Gain (LLG) which is an indication of how much "better" the solution is compared to a random solution. The higher your LLG the better the solution. An LLG of 0 or less means that your solution is no better or even worse than a random selection of atoms. The second thing to look at is the "Z-score" which shows you how many standard deviations your solution is above the mean, again the higher the better (above 5). In the above example I am using a homo-dimer as a search model so have found two strong solutions for the two possible orientations.
After checking your rotation function you need to scroll down to the bottom of the translation function section which should look something like this:
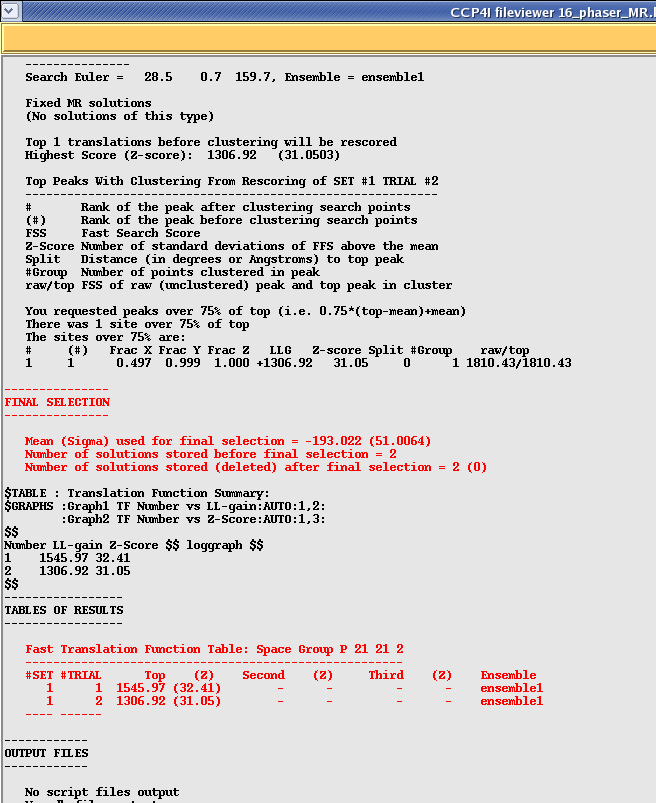
In this case the program has taken the top rotation function (SET 1) and found two possible solutions (with a high LLG and Z-score). According to the manual Z scores lower than 4 indicate no solution, 5 to 7 a possible solution and any higher than 7 a definite solution. So in this example 32 indicates a very strong solution!
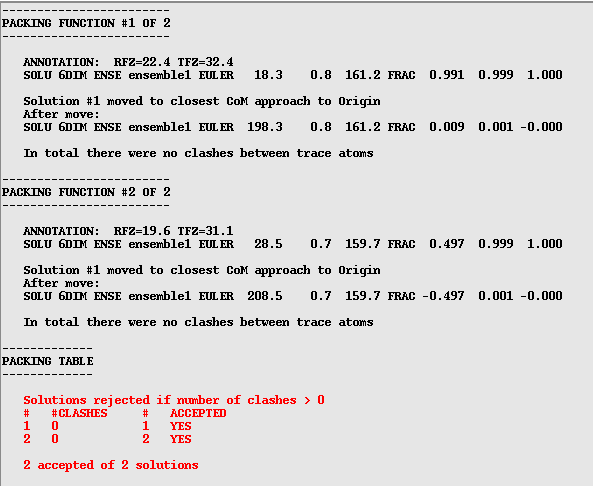
Next Phaser looks at the packing and works out whether there are any atomic clashes in the solution. In this example I have used the asymmetric unit as the search model so there will not be any clashes, however if you are asking the program to find more than one component there could well be clashes. A few clashes do not necessarily mean an incorrect solution as they could be due to flexible loops or disorder in the crystal. In default mode Phaser is quite strict about this and will reject all solutions with clashes. It is well worth checking this tables (see below) and see if there is a solution with only a few clashes (generally less than 10 or so). If this is the case, and Phaser has rejected the solution, you can re-run the job with the "Packing tolerance" (at the bottom of the "Search details" menu) set to a value higher than the clashes you want to allow (don't forget to select Packing tolerance by clicking on the square box!).
After the packing function phaser refines the model and then writes a pdb file for your strongest solution and an mtz file with the phases included (useful for model building in coot). It also gives you a .sol file with your solution represented as Euler angles that can be read back into Phaser if you need to modify or extend your search further. It tells you the name of these output files along with details of your solution right at the end of the file: